INTRODUCTION
Netrin-1 is a secreted protein that belongs to class of laminin-related hormones involved in neuronal axon guidance [1,2]. Netrin-1 is genetically conserved across nematode worms [2], fruit flies, frogs, mice, and humans. The reported netrin-1 receptors are more than a dozen, including deleted in colorectal cancer (DCC) [3,4], unc-5 netrin receptor (UNC5) orthologues (human UNC5A–D and rodent UNC5H1–4) [5], P53 [6], several integrins [7,8], immunoglobulin superfamily of transmembrane receptors (neogenin) [9], p75 neurotrophin receptor (p75 NTR) [10], rearranged during transfection (RET) [11], anaplastic lymphoma kinase (ALK) [12], and tropomyosin receptor kinase (TrkC) [13]. They mediate a variety of biological functions from neuronal activity in adulthood [14], synaptic plasticity [15,16] to inflammation [17,18]. DCC and UNC5H receptors are cleaved by major proteases of the apoptotic signal pathway, caspases, which are cysteine proteases that are activated at pH 7.4, cleaving intracellular proteins on the carboxyl side of an aspartate residue. Caspase family members involved in apoptosis can be divided into two groups: signal cascade initiator caspases (caspase-8 and -9) and effector caspases (caspase-3, -6, and -7). Their activation initiates a proteolytic cascade leading to the activation of the effector caspases, rapidly triggering the execution of apoptosis. UNC5B and DCC receptors are cleaved by effector caspase (caspase-3) at specific amino acid position D412 for UNC5B and D1290 for DCC [3,5].
The midbrain structure is mainly composed of dopaminergic neurons which related the Parkinson’s disease (PD) motor dysfunctions. Previously, it has been reported that the axons of dopaminergic neurons are guided by differential netrin-1 expression in the striatum (ST) via DCC receptor [19]. Also, netrin-1 and DCC, UNC5B or UNC5C continue to be expressed in the adult brain, in the brain regions where neurodegenerative diseases like PD affected [4,20]. The preferential and progressive degeneration of dopaminergic neurons of the substantia nigra pars compacta (SNpc) region in PD leads to motor defect, resulting in impaired quality of life in patients with PD. However, netrin-1 and DCC are highly expressed in healthy (non-PD) dopaminergic neurons of the SNpc specifically [21,22]. Nowadays, the cause of neurodegenerative diseases remains obscure and the current therapies only temporarily alleviate the symptoms. Interestingly, netrin-1 is substantially reduced in the brain during aging and in the PD patient brains [23]. Of note, we found that netrin-1 deficiency activates mammalian Ste20-like kinases 1 (Mst1) that triggers UNC5B proteolytic fragmentation via T428 phosphorylation, making dopaminergic neurons particularly vulnerable to degeneration [23]. Moreover, we provide the molecular mechanism of how CCAAT/enhancer binding protein β (C/EBPβ) suppresses netrin-1 transcriptional expression in PD, contributing to its pathologies via activating asparagine endopeptidase (AEP). Remarkably, this protease also cleaves UNC5C receptor, exacerbating PD pathogenesis.
NETRIN-1/RECEPTORS TUMORIGENESIS FUNCTIONS
Mounting evidence supports that axon guidance molecules are implicated in various human cancers. The expression of netrin-1 is markedly reduced or deficient in approximately 50% of brain tumors and neuroblastomas [24] and in prostate cancer [25]. Some of the reports show that inactivation of netrin-1 contributes to cancer development. However, conflicting reports also demonstrate that netrin-1 acts as an oncogene in tumorigenesis. For instance, netrin-1 protein expression level is extremely high in colorectal cancer [26]. Accumulative evidence supports that netrin-1 inversely regulates DCC- and UNC5H receptors-mediated apoptosis and p53-dependent apoptosis. Mazelin et al. [27] generated transgenic (Tg) mice that overexpressed netrin-1 in the gut. In this manuscript, s series of independent lines of Tg mice were obtained that had inserted a transgene containing netrin-1 complementary DNA under the gut-specific fatty acid-binding protein (Fabpl) (4x at -132) promoter18. Five of these lines (Tg-netrin-1/1 to 5) were analyzed. First of all, they shown netrin-1 (laminin-related secreted proteins, NTN1) expression of the transgene in the whole gastrointestinal (GI) tract. Moreover, tagged netrin-1 was detected by immunoblot and immunohistochemistry in epithelial villi and crypts isolated from the small intestine of Tg-netrin-1 mice. They address that inhibition of cell apoptosis by extrinsic increased expression of netrin-1 in mouse whole GI tract leads to the natural formation of hyperplastic and neoplastic lesions with these netrin-1 Tg mice. These results suggested that netrin-1 can promote colon cancer development, probably by regulating cell survival [27]. In addition to colorectal cancer [28,29], DCC deletion or mutation is also involved in testicular [30] and pancreatic cancers [31]. Hence, netrins and netrin receptors expression, gene deletion, and mutation are alternatively contributed to cancer’s progression in the different organs. In addition to tumorigenesis, netrin-1 and its receptors are also implicated in the neurodegenerative diseases.
MEDIATE PD PROGRESSION FROM THE ENTERIC NERVOUS SYSTEM TO THE CENTRAL NERVOUS SYSTEM VIA NETRIN-1, BRAIN-DERIVED NEUROTROPHIC FACTOR, AND C/EBPβ MOLECULES
Hereafter, to explore the potential roles of netrin-1 in substantia nigra (SN) dopaminergic neurons, we analyzed its expression in the adult mammalian SN using Allen Human Brain Atlas data bank. In the database bank, netrin-1 expression in the adult brain is globally low except in some structures of the brainstem. Remarkably, netrin-1 expression level is the highest one among the genes in the SN. Moreover, NCBI Gene Expression Omnibus (GEO) database (GDS2821 and GDS 3129) shows the significant decrease of netrin-1 expression levels in PD patient’s SN region; inversely, neuro-inflammation cytokines escalate transcription factor, C/EBPβ expression levels in overlapped brain region [32]. We have been identified the C/EBPβ transcription gene biological functions in neurodegenerative diseases [32-34].
The C/EBPβ acts brain-derived neurotrophic factor (BDNF)/ netrin-1 repressors upon neurotoxin and oxidative stress condition with interleukin 6 (IL-6), IL-1β, and tumor necrosis factor α cytokines overexpression. Moreover, C/EBPβ phosphorylation and activation are related the aging and neurodegenerative disease progression. Many research already identified C/EBPβ transcription factors in cytokines regulation. However, an unclear molecular mechanism remained in PD. Therefore, we investigated the C/EBPβ possible molecular function with netrin-1/BDNF in PD to explore the unknown molecular mechanism.
Neurotrophins are mainly synthesized in central nervous system (CNS) as well as in enteric nervous system (ENS). Neurotrophins including BDNF were decreased in many different brain regions during the aging process [35-37]. BDNF is expressed in the hippocampus, frontal cortex, midbrain, amygdala, hypothalamus, ST, pons, and medulla oblongata [38,39]. Moreover, BDNF is also expressed in the non-neuronal peripheral cells such as T and B lymphocytes, monocytes, vascular endothelial, smooth, and skeletal muscle cells [40-44]. BDNF promotes neuro-protection and neuro-regeneration. In PD animal models, BDNF promotes dopaminergic neuronal survival and improves neurotransmission and motor performance [45-47]. BDNF expression displays a similar pattern as netrin-1 in human and rodent. They both are agedependent decreased in the brain [33].
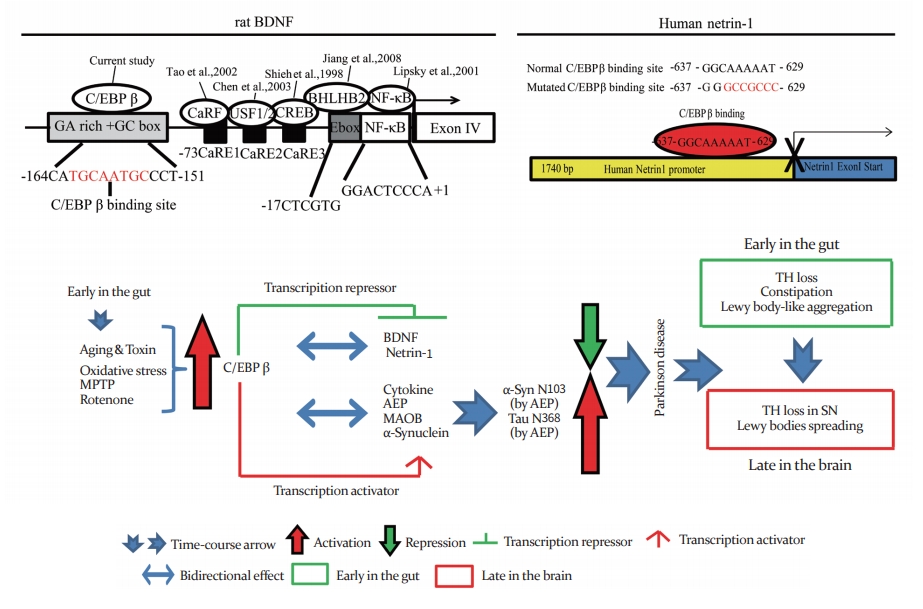
Our most recent study reveals that C/EBPβ is progressively escalated in the brain, inducing AEP mRNA expression, which mediates Alzheimer’s disease (AD) pathologies in animal models [34]. C/EBPβ belongs to the C/EBP family of transcription factors, binding to the CCAAT box of promoters and enhancer regions in numerous gene targets [48]. C/EBPβ is highly expressed in the intestine, liver, kidney, lungs, spleen, adipose tissue, pancreatic β-cells and in the CNS. C/EBPβ plays a key role in inflammation [49]. Promoters of many pro-inflammatory genes contain C/EBPβ consensus sequences [50]. Interestingly, the inflammation cytokines [34] and oxidative stress can induce C/EBPβ upregulation in dopaminergic neurons [51]. We found that both netrin-1 and BDNF were clearly diminished in PD patients’ gut and brain tissue samples, inversely coupled with robust C/EBPβ (T235) phosphorylation, a marker for its activation [33]. Conceivably, C/EBPβ might act as a repressor to suppress both BDNF and netrin-1 mRNA transcriptional expression. To test this possibility, we identified that C/EBPβ bound to the promoters containing BDNF exon IV and netrin-1 exon I in specific binding sequence regions in primary neurons and neuroblastoma cells, repressing their mRNA expression (Fig. 1). To further test this notion, we employed 3 months old C/EBPβ heterozygous mice and wild-type (WT) littermates. After 2 months of rotenone oral gavages (30 mg/kg), both C/EBPβ and p-C/EBPβ were evidently escalated in the gut and the brain of WT mice upon rotenone oral treatment. Accordingly, caspase-3 (apoptosis marker molecule) was activated, associated with UNC5B (netrin-1 receptor) and DCC (netrin-1 receptor) cleavage. Notably, tyrosine hydroxylase (TH)-positive dopaminergic neurons were evidently lost in the ENS in the gut, accompanied with extensive p-α-Syn S129 containing Lewy body-like inclusions. These events were diminished in C/EBPβ+/– mice. Consequently, C/EBPβ+/– mice displayed mild GI tract dysfunction and damage as compared to the prominent effects in WT mice group [33].
GI dysfunction is a prominent non-motor feature of PD. Before motor impairment, many PD patients suffer from delayed gastric emptying, constipation, and olfactory dysfunction [52-54]. Presumably, BDNF and netrin-1 reduction in the gut might account for the GI tract aberrant pathological effects in PD. To test this notion, we generated BDNF or netrin-1 gut conditional knock out mice. Remarkably, these mice exhibited dopaminergic neuronal loss in the ENS in the gut, constipation, inflammation, and motor dysfunctions [33], which resemble to the PD prodromal symptoms. These interesting observations may shed light into the potential molecular mechanisms mediating PD patient GI tract dysfunctions. Furthermore, we have reported that AEP-cleaved α-Syn N103 and Tau N368 truncates associate with each other much stronger than their full-length counterparts. The pre-formed fibrils between α-Syn N103 and Tau N368 spread from the gut along vagus nerve into the dorsal motor vagus nerves in the brainstem, from where they propagate into the SN, initiating PD pathologies and motor deficits [54]. Therefore, C/EBPβ activation that repressed both BDNF and netrin-1 expression levels in the gut, resulting in AEP activation and α-Syn N103 and Tau N368 fragmentation, culminating in dopaminergic neuronal loss and prodromal GI disorders (Fig. 1). The truncated fragments form the Lewy body-like inclusions in the gut and they translocate into the brain, spreading the PD pathologies from the ENS into the CNS. Hence, C/ EBPβ/AEP pathway provides an innovative molecular mechanism initiating PD pathogenesis.
NETRIN-1/DCC PAIR IN PD PATHOGENESIS VIA CASPASES
The pair of netrin-1/DCC are key molecules in the CNS mediating axonal and neuronal guidance. The expression of netrin-1 and DCC is maintained well in the adult brain; however, little is known about their roles in the mature neurons duringthe aging process. Interestingly, netrin-1 is highly expressed in the adult SN regions, which led us to investigate the role of the pair netrin-1/DCC in adult dopaminergic neuron’s fate.
Especially, SNpc dopamine (DA) neurons are particularly susceptible to degeneration. Both in human and mouse midbrain, that ventral SNpc DA neurons exhibited a stronger signal for DCC than other midbrain DA neurons (from dorsal SNpc and the ventral tegmental area) [21,55] suggesting that DCC or UNC5 homologues receptors and netrin-1 protein levels may be markers for DA neurons that are most vulnerable to degeneration.
DCC and UNC5B, belong to the class of dependence receptors, a functional family of receptors shown to be cleaved and to trigger apoptosis in settings of poor receptor availability [5,56]. During the aging, the DCC and UNC-5 homologues levels were escalated and these receptors are truncated by active caspase-3. Mehlen et al. [3] discovered that DCC induces apoptosis in the absence of the netrin-1, but apoptosis is blocked when engaged by netrin-1. Hence, DCC receptor acts as a ‘dependence receptor’ [57,58]. Dependence receptors generate survival signaling on their specific ligands and induce apoptosis when uncoupled by the netrin-1 ligand. DCC was also shown to be a caspase substrate, cleaved at Asp1290 by caspase-3. A point mutation at this site suppresses the proapoptotic effect of DCC completely, indicating that DCC must be cleaved to induce apoptosis. Furthermore, a specific domain known as a dependence domain has been mapped to the intracellular region of DCC, which lies upstream of the caspase cleavage site. The deletion of this domain is strong enough to destroy the pro-apoptotic effect of DCC. Although the sequence of this domain is not related to a known death domain or a caspase involvement domain that allow protein-protein interactions between a number of apoptosis trigging proteins, it seems likely that the dependence domain functions as a start to recruit and activate caspase-9 and caspase-3 [59]. DCC-induced apoptosis seems to be independent of both the mitochondrial apoptosis pathway and the death receptor/caspase-8 pathway. Instead, in the absence of netrin-1, DCC indirectly interacts with caspase-9 through an unknown protein and stimulates the formation of the caspaseactivating complex, resulting in activation of caspase-3 through caspase-9 without a requirement for the secrete of cytochrome c from the mitochondria or interaction with the scaffold protein apoptotic protease activating factor 1 (APAF1) [59].
Netrin-1/DCC pair tightly regulates cell survival and apoptosis. To explore netrin-1’s pathological role in PD pathologies, we employed netrin-1 fl/fl mice using Cre-virus injection into the SN region. We observed TH-positive neuronal loss and motor defects in netrin-1-depleted mice. A massive increase of terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) signals in TH-positive neurons in the SNpc demonstrated that conditional knockout of netrin-1 elicited death of dopaminergic neurons. Therefore, netrin-1 is the required element for DA neurons maintenance in adult animals. α-Syn phosphorylation (p-α-Syn S129), a PD pathological biomarker, and its aggregation were observed upon netrin-1 deprivation. The elevated DCC truncate and active caspase-3 were identified in the PD animal model. In primary neuronal cultures, neutralization of netrin-1 [60,61] escalated DCC or UNC5B protein levels and cleavage, associated with increased neuronal apoptosis. On the other hand, DCC or UNC5B expression levels triggered caspase’s activation in dopaminergic neurons. However, DCC might be a doubleedged sword for dopaminergic neurons depending on netrin-1 protein availability: supporting neuronal survival (and outgrowth) in the presence of netrin-1 and promoting cell death in conditions of trophic scarcity. As dopaminergic neurons are thought to be vulnerable to oxidative stress and inflammation due to their unique morphology and mitochondrial dysfunctions, DCC dual signaling could be a neuronal cell protective mechanism, eliminating damaged or disable neurons and protecting healthy neurons from additional harm. Our current research showed that netrin-1 silencing/deprivation were associated with increased α-Syn, monoamine oxidase-B (MAOB), C/EBPβ, DCC, UNC5B, caspase-3, α -Syn pS129, aggregates α-Syn, and AEP levels [33,62]. Moreover, netrin-1 expression and protein levels were progressively reduced in an age-dependent manner in the adult SN of alpha-synuclein gene (SNCA) Tg mice, emphasizing that there might be a link between α-Syn and netrin-1 levels. Injection of recombinant netrin-1 prevented behavioral motor dysfunctions in SNCA Tg mice with the reduced cleaved form of DCC, UNC5B during the caspase-3 and AEP enzyme activation [33,62].
NETRIN-1 DEPRIVATION ACTIVATES MST1 THAT PHOSPHORYLATES UNC5B, PROMOTING CELL DEATH
The MST1/2 (homologs of Drosophila Hippo) phosphorylates and activates large tumor suppressor 1/2 (LATS1/2) [63]. The Hippo pathway in both Drosophila and mammals regulates cell number by modulating cell death, cell proliferation, and cell differentiation. Orchestration of these processes plays a critical role in a variety of physiological and pathological conditions [64]. During development, an increase in cell number is an essential requirement for supporting organ maturation and functions; meanwhile, modest differentiation of multiple cell types warrants the appropriate functions of developed organs. In several studies, the Hippo (Mst1) pathway has been shown to induce cell death and differentiation. MST1/2 possesses additional functions in addition to regulating Hippo pathway components of LATS1/2 and yes-associated protein (YAP)/transcriptional coactivator with PDZbinding motif (TAZ). YAP and TAZ are transcriptional co-activators. When translocated into the nucleus, they regulate gene expression through interaction with TEA domain transcription factor 1–4, which are sequence-specific transcription factors that mediate the main transcriptional output of the Hippo pathway in mammalian cells [65]. The major physiological functions of YAP and TAZ are to induce cell survival and proliferation [66].
MST1 pathway is implicated in neuronal cell death. For example, Mst1/2 phosphorylates forkhead box protein O1 (FOXO1) to elicit its nuclear localization and transcription of genes promoting apoptosis in mammalian neurons [67]. Moreover, reduced Mst1 phosphorylation eliminates blood-brain barrier (BBB) damage, and neurobehavioral impairment and brain edema [68]. Previously, we showed that Akt phosphorylates MST1 on T387 and inhibits its proteolytic activation, blocking FOXO3 phosphorylation and nuclear translocation and stimulating cell survival [69]. Further, we have reported that netrin-1 induces the interaction of UNC5B receptor with the brain-specific GTPase phosphoinositide-3 kinase enhancer L (PIKE-L) [70]. This interaction stimulates PI3K/Akt activation, inhibits UNC5B receptor pro-apoptotic activity and enhances neuronal cell survival [71]. In our recent study, we reported that netrin-1 deprivation mediates MST1 activation in UNC5B receptor-dependent manner, and active Mst1 phosphorylates UNC5B, promoting its apoptotic cleavage through caspase-3 activation. Netrin-1 reduction stimulates dopaminergic neuronal loss in PD patient brain samples or mouse brain samples via activating MST1 that subsequently phosphorylates UNC5B on T428 residue, escalating its proteolytic cleavage and apoptosis [23]. Overexpression of un-phosphorylate UNC5B T428A mutant strongly suppressed netrin-1 deprivation-induced MST1 apoptotic activation, leading to repression of p-LATS/p-YAP pathway, which culminates in repression of caspase-3 activation and inhibition of dopaminergic apoptosis. Blockade of UNC5B T428 phosphorylation by MST1 not only inhibits UNC5B apoptotic cleavage and reduces its apoptotic activity but also diminishes MST1 apoptotic activation (Fig. 2). Hence, these findings support that UNC5B and MST1 mutually regulate each other apoptotic functions via physical interaction and phosphorylation, and netrin-1 modulates Hippo/MST1 signaling via its receptor UNC5B receptor [23].
MST1/2 are also involved in cellular oxidative stress responses [67,72]. On the other hand, YAP physically interacts with FOXO1 and activates FOXO1-mediated transcription of catalase and manganese superoxide dismutase (MnSOD) genes and thereafter reduces levels of reactive oxygen species (ROS) and ischemia/reperfusion-induced injury in the heart [73], suggesting a physiological role of YAP in ROS scavenge. Noticeably, MST1 is activated in neurodegenerative prion disease and neuro-inflammation mechanism [74]. Moreover, YAP has also been implicated in neurodegenerative diseases, for instance, Huntington’s disease. In transcriptional repression-induced atypical death of neurons, YAP full-length is reduced, triggering apoptosis [75]. Our recent studies shown that netrin-1 induces YAP levels via inhibiting Hippo/MST1 pathway and promotes cell survival are in alignment with the well-defined pro-survival functions of YAP [23,76].
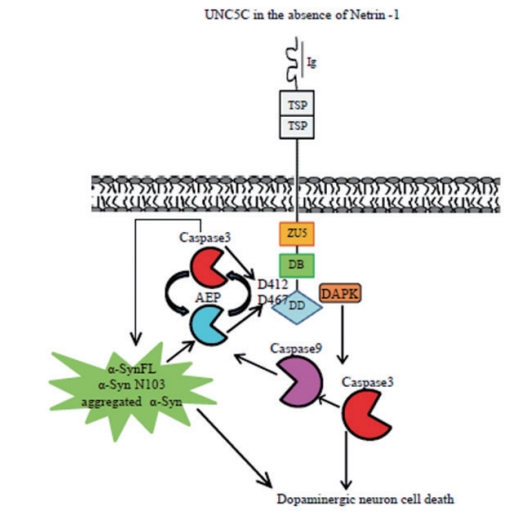
NETRIN-1 DEFICIENCY INDUCES AEP ACTIVATION AND UNC5C CLEAVAGE IN PD PATHOGENESIS
Netrin-1 receptors including DCC and UNC5Hs act as dependence receptors regulating neuronal apoptosis. Recently, we reported that AEP cleaves human α-Syn, promotes its aggregation and escalates its neurotoxicity, thus leading to dopaminergic neuronal death and motor dysfunctions in PD mouse model. AEP enzyme is activated and consequently cleaves human α-Syn at N103 residue in an age-dependent manner in SNCA Tg mice, and AEP is highly active in the SNpc regions in human brains with PD. Deletion of AEP from SNCA Tg mice alleviates dopaminergic neuronal loss and attenuates motor deficits [77]. Previous studies show that UNC5C expression is down-regulated in colorectal cancers and in several other cancers. This is also the case for UNC5A and UNC5B, mainly through promoter methylation [78-80]. Nevertheless, the tumor frequency is not increased in UN5C-deficient mice, suggesting that loss of UNC5C function is not sufficient to initiate tumorigenesis in mice. UNC5C serves as receptor in signaling pathways, recruiting other proteins into signaling complexes [81,82]. UNC5C is also widely expressed in the adult CNS neurons. It plays a critical role in the development of spinal accessory motor neurons [83]. Moreover, UNC5C functions as a chemotropic molecule in mediating axon growth and neuronal transplantation in neuronal development [84-87]. So far, six single nucleotide polymorphisms in UNC5C have been show to increase the risk of late-onset AD [88]. A recent mechanistic study showed that UNC5C T835M mutant in death domain induced neuronal cell apoptosis via serine/threonine death-associated protein kinase (DAPK1)/protein kinase D (PKD)/apoptosis signal-regulating kinase 1 (ASK1)/nicotinamide adenine dinucleotide phosphate (NADPH) oxidase/ caspases pathways [20]. In our most recent unpublished work, we show that netrin-1 reduction in PD patient brains or induced pluripotent stem cell (iPSC)-induced human neurons induces AEP activity, which robustly cleaves UNC5C at both N467 and N547 residues, augmenting TH-positive dopaminergic neuronal cell apoptosis. Moreover, we identified the netrin-1 is age-dependently repressed in the brains of SNCA Tg mice, associated with AEP activation and UNC5C fragmentation and dopaminergic neuronal loss. In alignment with these results, we observed that human α-Syn N103 truncation by AEP is highly escalated in PD brains and aged human SNCA Tg mouse brains, correlating with elevated p-α-Syn S129 activities. Consistently, overexpression of AEPtruncated UNC5C C-terminal fragment in the SN of human SNCA Tg mice strongly augments caspase-3 and AEP activation, accompanied with conspicuous dopaminergic neuronal cell loss, leading to α-Syn aggregation and motor dysfunctions. Therefore, netrin-1 deprivation in PD might induce both caspase-3 and AEP activation, which robustly cleave DCC and UNC5C that subsequently trigger cell death signals, culminating in dopaminergic neuronal cell apoptosis and defects (Fig. 3).
CONCLUSION
We have summarized the evidence that DCC, UNC5H family receptors, AEP, and C/EBPβ are crucial neuronal cell death factors in PD. Netrin-1 and BDNF are strongly reduced in the PD patient brains and gut tissues and rotenone-treated mice, and they also decline in SNCA Tg mice in an age-dependent manner. These innovative discoveries strongly support that netrin-1 and its receptors are potential targets for drug development for PD. Netrin-1 is mainly expressed in the brainstem and notably in the SN. These structures are predominantly affected by Lewy body pathology and neurodegeneration in PD. Netrin-1 deficiency triggers AEP upregulation via activating C/EBPβ transcription factor. In addition, C/EBPβ also acts as a repressor for both BDNF and netrin-1 via binding to their promoters [33]. C/EBPβ has different binding partners under different environmental conditions. C/EBPβ directly regulates IL-6, IL-1β, and nuclear factor-κB transcription under oxidative stress condition [49]. DA metabolite by MAOB, 3,4-dihydroxyphenylacetaldehyde (DOPAL), strongly induces AEP enzyme activity, whose mRNA transcription mediated by C/ EBPβ in the CNS [34,89]. Hence, our results support the notion that alleviating netrin-1 reduction via preventing neuroinflammation or oxidative stress-activated C/EBPβ/AEP repression might be the potential therapeutic strategy.