INTRODUCTION
Diabetes mellitus (DM) is a heterogeneous metabolic disorder characterized by hyperglycemia resulting from inadequate insulin secretion, resistance to insulin action, or both [1]. The major clinical presentation of DM is elevated blood glucose concentration; however, insulin resistance and/or insulin deficiency also cause abnormal lipid and protein metabolism and mineral and electrolyte disturbance. These uncontrolled metabolic disturbances lead to various microand macrovascular complications that are associated with lower quality of life and increased risk of morbidity and mortality.
Most DM patients are classified as type 1 diabetes mellitus (T1DM) or type 2 diabetes mellitus (T2DM). T1DM is characterized by insulin deficiency caused by autoimmune β cell destruction with markers of autoimmune destruction at the time of diagnosis in approximately 90% of patients [2,3]. Autoimmune responses against β cells are triggered by environmental factors in genetically predisposed individuals. Many genetic loci, including human leukocyte antigen (HLA) complex as well as non-HLA genes including insulin (INS), protein tyrosine phosphatase non-receptor type 22 (PTPN22), cytotoxic T-lymphocyte associated protein 4 (CTLA4), are associated with T1DM with intermediate or low effect size [4]. Meanwhile, the key defects in T2DM are progressive dysfunction in β cell insulin secretion and insulin resistance [5,6]. T2DM is a multifactorial disease that is associated with environmental, epigenetic, and genetic risk factors. More than 40 genes have been identified to increase the risk of T2DM, although the effect sizes are small [5,7].
Approximately 1% to 6% of pediatric diabetes cases are caused by mutations in one of more than 40 genes that are associated with β cell function or insulin action and are termed monogenic diabetes [1,8]. The main categories of monogenic diabetes include neonatal diabetes mellitus (NDM), maturity-onset diabetes of the young (MODY), and syndromic diabetes. It is important that a precise etiologic classification is made in order to determine therapy, the prognosis of complications, and genetic diagnosis; however, misdiagnosis is frequent since the phenotypes are overlapped [8]. In this review, we introduce the types of monogenic diabetes, clinical implications of diagnosis, and strategies for approaching patients suspected of monogenic diabetes.
TYPES OF MONOGENIC DIABETES
Neonatal diabetes mellitus
NDM is characterized by mild-to-severe hyperglycemia observed within 6 months of age that is associated with partial or complete insulin deficiency and can be classified as transient NDM (TNDM) or permanent NDM (PNDM) based on the clinical course [8]. The estimated incidence of NDM ranges from 1:90,000 to 1:260,000 among live births [9-11]. More than 30 genes have been identified in approximately 80% of NDM cases [12]. The common genetic causes and clinical features are summarized in Table 1. Clinical manifestations of NDM include hyperglycemia, glycosuria, polyuria, severe dehydration, and failure to thrive [13,14]. Also, infants with NDM are born small for gestational age or with intrauterine growth retardation, reflecting insulin deficiency in utero that underlines the importance of insulin in fetal growth.
PNDM that does not achieve significant treatment remission accounts for half of NDM cases. The majority of PNDM cases are caused by genes encoding KATP channel genes (potassium inwardly rectifying channel subfamily J member 11 [KCNJ11] and adenosine triphosphate [ATP] binding cassette subfamily C member 8 [ABCC8]) and genes encoding insulin itself (INS) [12]. The KATP channel consists of four subunits of the inward-rectifying potassium channel 6.2 (Kir6.2, encoded by KCNJ11) and four regulatory subunits of sulfonylurea receptor 1 (SUR1, encoded by ABCC8) and regulates insulin secretion in response to an increase in the ATP to adenosine diphosphate ratio, which makes the KATP channel close. The closure of the KATP channel leads to intracellular K+ accumulation and results in cell membrane depolarization, opening of voltage-gated Ca2+ channels, influx of Ca2+, and secretion of insulin [15]. Activating variants of KCNJ11 and ABCC8 keep the KATP channel in an open state regardless of ATP generation, leading to impaired insulin secretion [16]. Since KATP channels are expressed in the muscles and brain, approximately 20% of patients with KCNJ11 mutations are presented with muscular weakness, developmental delay, and early-onset epilepsy, in addition to NDM. Patients with KATP -NDM can be treated successfully with high-dose sulfonylureas, which are proven safe and effective for long-term glycemic control with only minimal hypoglycemia [17,18].
The second most common cause of PNDM is mutation in the preproinsulin gene (INS), which leads to misfolding proinsulin. The unfolded proinsulin results in loss of normal trafficking and accumulates in the endoplasmic reticulum (ER), leading to severe ER stress and apoptosis of β cells [19,20]. Two causative genes of MODY, GCK (encoding glucokinase) and Pancreatic And Duodenal Homeobox 1 (PDX1) (encoding the islet formation transcription factor) can be a rare cause of PNDM when in the homozygous or compound heterozygous state [21,22].
TNDM typically presents within a few weeks after birth and resolves spontaneously within 18 months of age, although it may relapse later in life [23]. Approximately 70% of TNDM cases are due to overexpression of the paternally imprinted genes at 6q24, including PLAG1 like zinc finger 1 (PLAGL1) and hydatidiform mole associated and imprinted (HYMAI), which results from paternally inherited duplication of 6q24, uniparental paternal disomy of the entire chromosome 6 or the segmental region that includes 6q24, or maternal hypomethylation of the differentially methylated region at 6q24 [14,24]. A transgenic mouse model of TNDM suggested that overexpression of PLAGL1/HYMAI, which has antiproliferative properties, leads to underdevelopment of the pancreas as well as impaired β cell function [25,26]. Compensatory increases in β cell number for reduced insulin synthesis and secretion restores normal fasting glucose, although hyperglycemia is sustained after the glucose challenge. Patients with 6q24-related TNDM are sensitive to insulin and achieve catch-up growth within a few weeks of treatment. Even after remission during infancy, it is important to be aware of the possibility of relapse around the time of adolescence [27].
The remaining causes of TNDM are mildly activating mutations in the KCNJ11 (Kir6.2) and ABCC8 (SUR1) genes, which encode KATP channels. In addition, it can, rarely, be caused by mutations in INS (encoding the preproinsulin), hepatocyte nuclear factor 1β (HNF1B; transcription factor), and solute carrier family 2 member 2 (SLC2A2; encoding the GLUT2 transporter) (encoding the GLUT2 transporter). These mutations are functionally less severe than those causing PNDM.
Maturity-onset diabetes of the young
MODY is characterized by autosomal dominantly-inherited and non-autoimmune DM, which usually presents at a young age with some additional features. The estimated prevalence ranges from 1:10,000 to 1:23,000, varies by ethnic and racial group, between children and adults [28-30]. The majority of MODY cases are caused by three genes (GCK, hepatic nuclear factor 1 alpha [HNF1A], and HNF4A), although at least 14 genes have causative genes identified for MODY or a MODY-like phenotype (Table 2) [8].
Glucokinase, encoded by GCK, initiates glycolysis by facilitating the phosphorylation of glucose to glucose-6-phosphate and acts as a glucose sensor in the pancreatic β cell. The inactivation mutation of GCK increases the set point of glucosestimulated insulin secretion [31,32]. Most patients with GCK-MODY have mild fasting asymptomatic hyperglycemia within the prediabetic range and elevated hemoglobin A1c levels which usually do not exceed 7.5% [33,34]. GCK-MODY has a low risk of diabetes-related micro- and macrovascular complications, and treatment is not generally required in these patients except under pregnancy conditions [35,36]. An unaffected fetus born to a mother with GCK-MODY is associated with increased risk of the perinatal complications of gestational DM due to excessive insulin secretion in response to maternal hyperglycemia, while the affected fetus will have normal birth because an altered set point is shared. A fetus with paternally inherited GCK-MODY born to an unaffected mother is at risk for low birth weight [37].
The common symptomatic MODY that requires treatment is caused by mutations in HNF1A or HNF4A, which account for approximately 30% to 65% of all MODY cases [29,38,39]. These genes encode pancreatic transcription factors that are important for the transcription of genes related to β cell development and insulin secretion. The diagnosis of DM in patients with HNF1A/HNF4A-MODY is made in adolescence or young adulthood, related to the location of the mutations and exposure to maternal diabetes in utero [40-42]. Transient hyperinsulinemic hypoglycemia and large for gestational age can be observed in some patients with HNF1A/HNF4A-MODY [43,44]. The frequency of micro- and macrovascular complications is similar to that of patients with T1DM and T2DM [45]. Patients with HNF1A/HNF4A-MODY are sensitive to lowdose sulfonylureas and achieve better glycemic control than with insulin or metformin [46,47].
Syndromic diabetes mellitus
A syndromic DM should be considered in any child or adolescent with diabetes associated with multi-system extrapancreatic features [48]. The Online Mendelian Inheritance in Man website (www.ncbi.nlm.nih.gov/omim or www.omim.org) can help with clinical features to determine whether the gene for a particular syndrome has been defined and hence whether molecular genetic testing is available.
Syndromic NDM constitutes a relatively small fraction of NDM [12]. The spectrum of a genetic disorder can result in abnormalities in pancreas formation and function and may have accompanying severe congenital anomalies, such as intestinal and biliary atresia (regulatory factor X6 [RFX6]), congenital heart defects (GATA binding protein 6 [GATA6] or GATA binding protein 4 [GATA4]), brain malformations (pancreas associated transcription factor 1a [PTF1A], neuronal differentiation 1 [NEUROD1], immediate early response 3 interacting protein 1 [IER3IP1], motor neuron and pancreas homeobox 1 [MNX1], NK2 homeobox 2 [NKX2.2]), or skeletal dysplasia (eukaryotic translation initiation factor 2 alpha kinase 3 [EIF2AK3]), which are difficult to recognize during the neonatal period. In addition to mutations in forkhead box P3 (FOXP3), which is responsible for the IPEX syndrome (immune dysregulation, polyendocrinopathy, enteropathy, X-linked), several additional genes (signal transducer and activator of transcription 1 [STAT1], STAT3, LPS responsive beige-like anchor protein [LRBA], interleukin 2 receptor subunit alpha [IL2RA]) have now been described to have mutations causing infancy-onset syndromic diabetes, along with other autoimmune disorders that result from dysfunction of immune regulation [49-52].
Wolfram syndrome (WFS), also known as diabetes insipidus, DM, optic atrophy, and deafness syndrome, is diabetes associated with progressive optic atrophy below 16 years of age, although it may present anytime from early infancy [53]. Other typical clinical features including sensorineural deafness, central diabetes insipidus, urinary tract dysfunction, and neurological symptoms that develop later in life with variable order even within the same family [54-56]. At least 90% of patients have a recessive mutation in the WFS1 gene (Wolfram syndrome 1, WS1). Recently, a presentation similar to WS1 in many WFS1 mutation-negative patients was linked to a variant in CDGSH iron-sulfur domain 2 (CISD2) that is named Wolfram syndrome 2 (WS2). Clinical features of this variant do not include diabetes insipidus, instead, patients present with peptic ulcer bleeding and defective platelet aggregation [57].
Renal cysts and diabetes (RCAD) syndrome (HNF1B-MODY) is a rare subtype of familial diabetes, and heterozygous mutations in HNF1B rarely present with isolated diabetes [58]. Renal developmental disorders, mostly renal cysts and renal dysplasia, are present in almost all patients with HNF1B mutations or gene deletions even in the absence of diabetes [59,60]. Genital tract malformations, hyperuricemia, gout, and abnormal liver function test results can also occur [58]. Diabetes develops later, usually during adolescence or early adulthood, although TNDM has been reported in a few cases [61-64]. In addition to insulin deficiency related to pancreatic hypoplasia, patients also show some degree of hepatic insulin resistance and require early insulin therapy [65-67]. Also, lower exocrine pancreatic function with reduced fecal elastase presents in mutation carriers [68]. Therefore, the phenotype of RCAD patients is highly variable even within families sharing the same HNF1B mutation, and RCAD should be considered not only in the diabetes clinic but also in other clinics including nephrology, urology, and gynecology. In patients with renal cysts, imaging of the pancreas and fecal elastase should be considered. The absence of the pancreatic body and/or tail and abnormal fecal elastase indicates HNF1B-MODY [68]. One-third to two-thirds of cases showed de novo mutations and deletions of this gene [59,60].
Mitochondrial diabetes, also known as maternally inherited diabetes and deafness (MIDD), is caused by pathogenic variants in mitochondrial DNA, mostly tRNA variant m.3243A>G. Diabetes due to mitochondrial mutations and deletions is rarely seen in children and adolescents, and the majority of cases develop in young or middle-aged adults [69]. However, some cases have been reported in adolescents with a high degree of heteroplasmy [69-71]. Mitochondrial diabetes should be suspected in patients presenting with diabetes and sensorineural hearing loss inherited from the mother’s side or diabetes and progressive external ophthalmoplegia. Other clinical features such as macular pattern dystrophy, nephropathy, and neurological symptoms are more common in rarer forms of mitochondrial diabetes than classical forms [72]. Interestingly, the same m.3243A> G mutation also causes a much more severe clinical syndrome known as myopathy, encephalopathy, lactic acidosis, and stroke (MELAS) [73]. Patients with mitochondrial diabetes may respond initially to diet or oral hypoglycemic agents but often require insulin treatment within months or years. Metformin should be avoided as it interferes with mitochondrial function and may trigger episodes of lactic acidosis [74].
CLINICAL IMPLICATIONS OF THE DIAGNOSIS OF MONOGENIC DIABETES MELLITUS
Genetic testing in patients suspected of monogenic diabetes has several clinical implications. Establishing a definite diagnosis is important for effective management based on the pathophysiology of diabetes. For example, avoiding insulin and sulfonylurea treatment can achieve successful glycemic control in the NDM caused by the activating mutations in KCNJ11 or ABCC8. Furthermore, genetic testing provides a better understanding of long-term prognosis and enables an avoidance of unnecessary treatment for diagnoses such a GCK-MODY, which has a low risk of diabetes-related complications and does not require treatment. Also, genetic diagnosis enables early evaluation and follow-up of extra-pancreatic manifestations of syndromic diabetes. For example, MODY caused by HNF1B mutations or 17q12 microdeletions is related to structural abnormalities in the kidney and urinary tract, hypomagnesemia, and abnormal liver function [68]. In the same way, DM caused by WFS1 has been associated with diabetes insipidus, optic atrophy, and deafness; CISD2 with optic atrophy, peptic ulcer bleeding, and defective platelet aggregation; and mitochondrial diabetes with deafness, nephropathy, and neurological symptoms [57,72,75].
DIAGNOSTIC APPROACH AND INTERPRETATION OF GENETIC TESTING
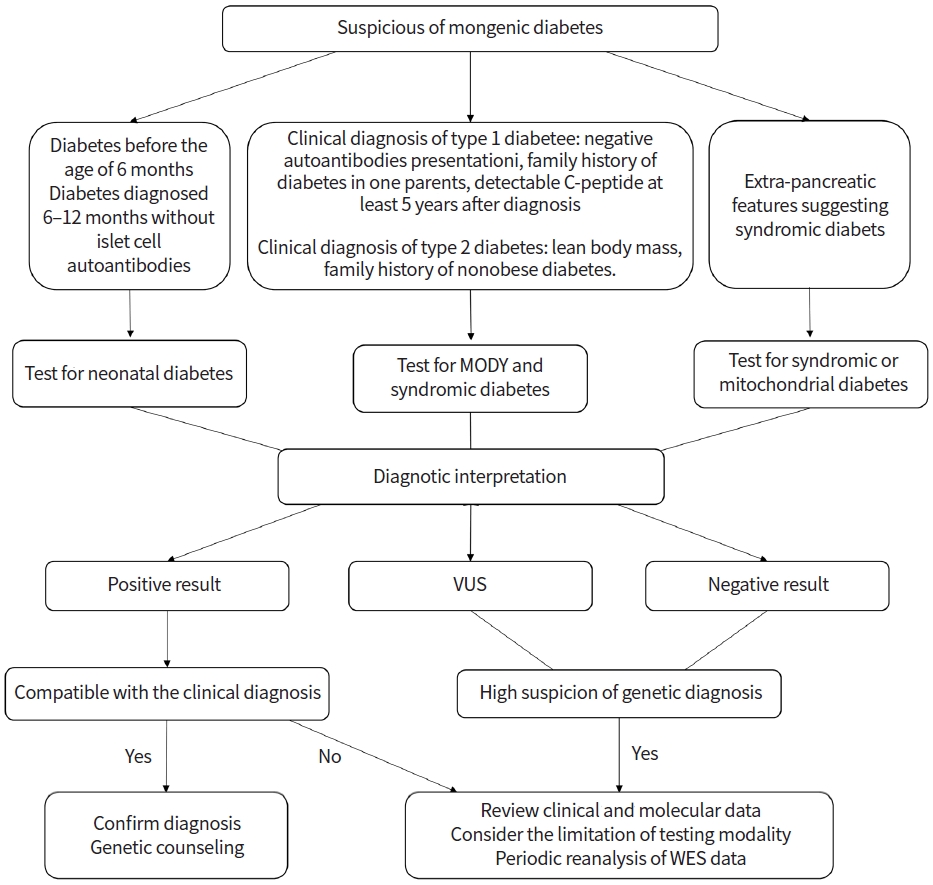
There are limited practical guidelines for standardized diagnostic approaches and genetic testing for monogenic diabetes. Fig. 1 summarizes the proposed diagnostic approach for monogenic diabetes. The International Society for Pediatric and Adolescent Diabetes (ISPAD) has recommended genetic testing for patients diagnosed with DM before 6 months of age and patients diagnosed with DM between the age of 6 and 12 months without islet autoantibodies [8].
Approximately 80% of monogenic diabetes cases in pediatric and young adults are misdiagnosed as T1DM or T2DM; monogenic diabetes should be suspected by clinicians for patients with a certain combination of clinical and laboratory results [28,76]. Monogenic DM should be considered in T1DM patients who had negative autoantibodies at presentation, family history of diabetes in one parent, and preserved insulin secretion with detectable C-peptide at least 5 years after the presentation [8]. The features that should be suspected of monogenic diabetes in T2DM include lean body mass, lack of evidence of insulin resistance and/or other features of metabolic syndrome, family history of nonobese diabetes, and unusual fat distribution. Syndromic diabetes should be considered for DM cases with extra-pancreatic features such as deafness or neurological features.
As several causative genes have been identified in monogenic diabetes, the targeted gene panel, which consists of the genes considered to be associated with monogenic diabetes, is a rapid and cost-effective test. This targeted approach simplifies the interpretation of the results and minimizes the potential detection of secondary and incidental findings. Chromosomal microarray analysis also could be considered in patients with distinct features of a contiguous gene deletion such as 17q12 deletion syndrome. If the genetic cause is not detected with the targeted panel test, whole-exome sequencing (WES) and whole-genome sequencing (WGS) could be considered. WES and WGS are relatively unbiased genome-wide tests that cover protein-coding regions. WES and WGS allow for the discovery of new causative genes of monogenic diabetes. However, there is an issue of interpretation of results that include a large number of sequence variations and incidental findings. According to the recommendations in the American College of Medical Genetics and Genomics guidelines, the variants are assessed using various types of evidence and are classified into one of five categories: benign, likely benign, variant of uncertain significance, likely pathogenic, and pathogenic [77]. Incidental findings that are important to health and are medically actionable, such as those of cancer or cardiomyopathy, should be assessed and reported in patients undergoing WES or WGS [78].
Appropriate genetic counseling should be provided, and documentation of consent should be obtained when genetic testing is required [79]. This discussion contains the basic explanation of genetic disease and testing modalities; expected testing results; the advantages, disadvantages, risks, and limitations of testing; and testing cost. The possibility of incidental findings of medically actionable genes should be informed. Genetic counseling should be also provided after testing. The interpretation of test results, inheritance pattern, and risk of recurrence should be included. Also, information on available management, long-term prognosis, the need for further evaluation, and implications for the family should be provided.
CONCLUSION
Definite genetic diagnosis of monogenic diabetes is important for optimized treatment, notably in patients requiring no treatment (GCK-MODY), switching from insulin or metformin to sulfonylurea (HNF1A-MODY and HNF4A-MODY), and switching from insulin to high-dose sulfonylurea (KATP-NDM). In addition, a definite genetic diagnosis provides a better understanding of clinical course and familial recurrence risk. With recent advances in genetic testing technology, the capacity of genetic diagnosis has been increased with decreased cost. It is important for clinicians to know when and how to apply genetic testing in patients with DM. Therefore, careful, precise, and comprehensive assessment by clinicians is essential for suspicion of monogenic diabetes, encompassing patient and family history, physical examination, biochemical testing, and radiologic testing.