INTRODUCTION
Inflammatory bowel diseases (IBDs), primarily Crohn’s disease (CD) and ulcerative colitis (UC), are multifactorial diseases that develop aberrant immune responses in the intestines involving environmental factors, genetic variants, and intestinal dysbiosis [1-3]. Considering the disease characteristics of chronic relapsing inflammation with progressive complications, lifelong treatment strategies to prevent complications and maintain long-term remission are required. The development of treatment options has revolutionized the overall prognosis of IBD. Conventionally, 5-aminosalicylic acids (5-ASAs) are prescribed as first-line agents in patients with mild to moderate disease; glucocorticosteroids are prescribed for remission induction in moderate to severe disease and thiopurines for maintaining remission [4,5]. In addition, anti-tumor necrosis factor-α (anti-TNFα) agents have been shown to be effective for both inducing remission and maintenance in conventional therapy refractory disease [4], and anti-TNFα therapy has replaced treatment strategy from symptom control to mucosal healing [6-8]. However, a particular group of patients still experience therapeutic failure or adverse events. The high inter-patient variability in therapy is associated with clinical factors, including age, disease behavior, and disease duration [9,10]. However, they attribute only a small proportion of interindividual variability, and currently, genetic variation has been focused on the implementation of personalized IBD treatment. Pharmacogenetics evaluates the associations between specific genetic variations and drug responses to optimize therapeutic efficacy and minimize toxicity [9]. Recently, extreme phenotype selection strategy has been adopted as a methodology for pharmacogenetic analysis [11,12]. This strategy has identified relevant biological factors with effectiveness by reducing the sample size required for molecular studies, and interpreting the large amount of data generated by high-throughput techniques [13,14]. In this article, we review the identified pharmacogenetics of currently prescribed drugs, focusing on 5-ASAs, glucocorticosteroids, thiopurines, and anti-TNFα.
5-AMINOSALICYLIC ACIDS
5-ASA is the first-line treatment for the induction and maintenance of IBD remission [4,15,16]. 5-ASA acts locally on the colonic mucosa and reduces inflammation by activating nuclear receptors involved in the control of inflammation, γ-form peroxisome proliferator-activated receptors (PPAR-γ) which inhibit several target genes, including nuclear factor kappa B (NFκB), leukotrienes, prostaglandins, and interleukin (IL)-1 [17,18]. Thus, mucosal concentrations of 5-ASA are important to intensify drug effects [15]. In the intestinal mucosa, 5-ASA is poorly absorbed into the systemic circulation and rapidly N-acetylated mainly by the enzyme N-acetyl-transferase 1 (NAT1), and to a small degree by NAT2 [19]. Genetic polymorphisms in these enzymes lead to rapid or slow acetylation, and such variants are found in over 50% of Caucasians as slow acetylators [20].
However, NAT genotypes are unlikely to play a role in predicting 5-ASA responses. In 78 UC patients, the genetic variations of NAT1 (NAT1*3, *4, *10, and *11 allele) were not associated with response or side effects to mesalamine [21]. In addition, in a population-based cohort of patients with UC, NAT1, and NAT2 genotypes did not predict clinical response or toxicity to 5-ASA [22]. Recently, a study evaluated the association between 5-ASA concentration, 5-ASA formulation, NAT genotype, and microbiome in quiescent UC patients receiving 5-ASA monotherapy. Mezavant (Shire Pharmaceutical Contracts Ltd., in partnership with Cosmo SpA, Milan, Italy) provided higher 5-ASA mucosal concentration than Pentasa (Ferring Pharmaceuticals, Saint-Prex, Switzerland) (2.39 ng/mg vs. 0.57 ng/mg, P= 0.033). Although patients with NAT1 slow acetylators had higher 5-ASA serum concentrations, no influence was found in terms of mucosal 5-ASA concentration. Interestingly, mucosal 5-ASA concentration was positively related with gut microbial diversity and composition [23].
PPAR-γ gene expression, a key mediator of inflammation, was decreased in the active UC group compared with that in the UC in remission (P= 0.001) and control groups (P= 0.001) [24]. Increased expression of the PPAR-γ gene was associated with a milder clinical course (odds ratio [OR], 0.05; P≤ 0.001) [24] and milder endoscopic disease activity in active UC [25].
Considering the need for long-term maintenance of 5-ASA treatment, a genome-wide association study was performed to identify the risk factors associated with rare idiosyncratic nephrotoxicity in IBD patients. In this study, the median time for renal injury was 3 years after 5-ASA administration. The strongest association signal was in the human leukocyte antigen (HLA) region (rs3135349, OR, 2.04; P= 1× 10−7), and the top single nucleotide polymorphism (SNP) after dedicated HLA imputation was rs3135356 (OR, 2.0; P= 1× 10−7). When limited to biopsy-proven interstitial nephritis cases, the most associated HLA allele was HLA-DRB1*03:01 (OR, 2.76; P= 5× 10−7) [26]. However, its clinical utility is not recommended because of its high frequency of risk alleles and low frequency of nephrotoxicity [9].
Recently, a novel genetic factor associated with mesalamine allergy has been reported. Mesalamine allergy, which is characterized by high fever, worsening diarrhea, or bloody stool makes it difficult to distinguish from exacerbation of IBD [27]. These symptoms are more common in UC than those in CD, with varying incidence from 2.1% to 24% [16,27,28]. Mesalamine allergy is considered a type IV allergic reaction mediated by antigens and T cell-recognizing antigens (especially T helper 1 cells) [27]. Although the drug-induced lymphocyte stimulation test has been proposed as a diagnostic test, the positive rate of mesalamine intolerance was 24%, suggesting the presence of multiple mechanisms besides allergic reaction [28]. In an effort to identify the genetic background of mesalamine intolerance, the Japanese study team identified rs144384547 (upstream of regulator of G-protein signaling 17 [RGS17]) to be significantly associated with mesalamine-induced fever and diarrhea (OR, 11.2; P= 7.21e-09) with an estimated heritability of 25.4% [29]. Although information regarding the role of RGS17 in IBD or mesalamine pharmacogenetics is unavailable, RGS17 has been reported as a negative modulator of G-protein-coupled receptor signals in several types of cancer [30]. The combined genetic and clinical prediction models which could overcome the limitation of single genetic polymorphism analysis, yielded a higher area under the curve than the polygenic risk score or clinical model alone (area under the curve, 0.89; sensitivity, 71.4%; specificity, 90.8%) [29]. Pharmacogenetic candidates for the management of inflammatory bowel disease are summarized in Table 1.
GLUCOCORTICOIDS
Systemic glucocorticoids are the first-line conventional drugs for inducing remission of moderate to severe exacerbation of IBD [4]. Potent and rapid anti-inflammatory effects follow binding of the intracellular glucocorticoid receptor and inhibition of T cell activation and cytokine secretion [31]. However, approximately 20% of patients are resistant to glucocorticoids, and 30% to 40% of patients acquire dependency [32]. In a population-based study of patients with IBD, immediate outcomes for CD and UC were as follows: complete remission, 58% and 54%; partial remission, 26% and 30%; and no response, 16% and 16%. One year after the first course of treatment with corticosteroids, 32% of CD patients and 49% of UC patients had prolonged responses, whereas 28% of CD patients and 22% of UC patients showed corticosteroid dependence [33].
Glucocorticoids passively diffuse across the plasma membrane and activate a cytosolic glucocorticoid receptor which leads to repression or induction of the expression of several inflammatory genes [34]. As the mechanism of action of glucocorticoids is complex, glucocorticoid resistance might occur at several distinct molecular levels [35].
The glucocorticoid receptor variant is the most well-researched candidate involved in glucocorticoid pharmacogenetics. Polymorphisms of the glucocorticoid receptor gene nuclear receptor subfamily 3, group C, member 1 (NR3C1) impair the formation of the glucocorticoid-glucocorticoid receptor complex and alters transactivation or transrepression processes. In patients with steroid-resistant UC, the expression level of glucocorticoid mRNA in the intestinal mucosa is decreased [36]. To date, three polymorphisms, TthIII1, ER22,/,23EK, and GR-9β, are known to be associated with reduced sensitivity to endogenous or exogenous glucocorticoids. However, the role of these polymorphisms in IBD treatment is not well established. In 119 pediatric patients with IBD, no association was observed between ER22/23EK polymorphisms and glucocorticoid response [37].
Among SNPs of the NR3C1 gene, the Bcl1 and N363S polymorphisms have been reported to have increased glucocorticoid sensitivity [34]. Bcl1 polymorphism consists of a C> G substitution, 646 nucleotides downstream from exon 2 [10,38], and heterozygous and homozygous carriers of the G allele showed hypersensitivity to glucocorticoids [38]. In a pediatric IBD study, a higher frequency of Bcl1 mutation was associated with glucocorticoid hypersensitivity in the glucocorticoid response group than that in the dependent group (OR, 3.61; 95% confidence interval [CI], 1.44 to 9.01; P0.006/0.030) [37]. Although the N363S polymorphism was found to have an influence on increased sensitivity to glucocorticoids in vivo [39], so far, there has been no association with clinical response to glucocorticoids in IBD patients [37]. However, a meta-analysis reported that glucocorticoid receptor gene polymorphisms, including Bcl1, ER22/23EK, and N363S were not associated with glucocorticoid resistance in IBD [40], and further well-designed cohort studies are needed.
Variants in the principal effectors of glucocorticoids are also considered to play a role in interindividual differences in clinical drug response. The most well-identified candidate is the drug efflux pump P-glycoprotein (Pgp) encoded by the multidrug resistance gene 1 (MDR1 gene)/ATP binding cassette subfamily B member 1 (ABCB1) gene [34,41]. Pgp, expressed on the apical surface of lymphocytes and intestinal epithelial cells [1], actively extrudes structurally unrelated substances from cells, resulting in reduced intracellular concentrations of glucocorticoids [10]. Among more than 50 polymorphisms described in the MDR1 gene, a non-coding SNP in exon 26 (C3435T) and tri-allelic G2667T/A have been considered as factors for IBD susceptibility [42-44] and response to glucocorticoids [42,45,46]. However, the results of these studies are conflicting, and further international efforts or meta-analyses are required.
Pro-inflammatory cytokines involved in the pathogenesis of IBD have been suggested to interfere with glucocorticoid receptor signaling [47]. The 308G >A (rs1800629) polymorphism of the TNFα gene is associated with steroid dependency in CD [48]. IL-1β polymorphism involved in an inflammatory cascade had no correlation with steroid response, whereas polymorphism of caspase-1 showed a low response to corticosteroids [49].
A recent study analyzed 21 genes associated with glucocorticoid receptors, transporters, cytokine genes, chaperones/co-chaperones, and kinases using the amplicon next-generation sequencing method and reported four genes as pharmacogenetic biomarkers to determine variable reactions of glucocorticoids. In this study, the c.1088A> G polymorphism in the NR3C1 gene was associated with glucocorticoid resistance (P= 0.002) and variant c.241+ 6A> G of the FK506 binding protein 5 (FKBP5) gene with glucocorticoid sensitivity (P = 0.040). In CD, the change c.2685 +49T >C of the ABCB1 gene was linked to glucocorticoid resistance (P=0.034), whereas the deletion c.306-7delT in the MAPK14 gene was linked to an adverse therapeutic effect (dependency and resistance, P=0.041) in UC [31].
THIOPURINES
Thiopurines have been the mainstay treatment for IBD for the maintenance of long-term remission and steroid-sparing agents [50-52]. They also reduce the risk of anti-drug antibodies and increase the efficacy of anti-TNFα agents [53]. Oral administration and relatively lower costs are their advantages in clinical usage [54]. However, variability in efficacy and toxicity related to inter-individual pharmacokinetic differences and genetic polymorphisms can result in drug discontinuation. Approximately 25% of patients have to stop thiopurine due to adverse reactions including myelosuppression, hepatotoxicity, gastric intolerance, and increased risk of malignancy [55].
Myelosuppression is the most common and serious dosedependent adverse effect of thiopurines, which further cause infection and mortality [54,56]. Leukopenia, the most common form of thiopurine-induced myelosuppression, develops at any time during treatment, ranging from 12 days [57] to 27 years [58]. Severe cases usually occur within the first month [56]. The incidence of leukopenia is higher in East Asian IBD populations including Koreans (31.2% to 39.6%) [59,60], Chinese (15.6%) [61], and Japanese (20.8%) [62] than that in Caucasians (5%) [63,64], which reflects the different genetic contributions of thiopurine metabolism.
Thiopurine drugs, azathioprine and its analog, 6-mercaptopurine, are prodrugs that require multiple enzymatic processes to become active metabolites [65]. The synthetic pathway of the active metabolites is in competition with inactivation pathways mediated by xanthine oxidase or thiopurine S-methyltransferase (TPMT). Thus, patients with the TPMT mutation have an increased risk of thiopurine-induced leukopenia [66], and the U.S. Food and Drug Administration recommends pretreatment TPMT test [51]. Low TPMT activity is related to increased 6-thioguanine nucleotide levels by increasing inosine monophosphate dehydrogenase activity, which subsequently leads to myelotoxicity. The major alleles accounting for TPMT deficiency were TPMT*2, TPMT*3A, and TPMT*3C [67]. In Caucasians, 0.3% are homozygous for low enzyme activity (complete deficiency), 11% are heterozygous (partial deficiency), and 90% are homozygous for high enzyme activity (high activity) [68]. However, Asians have a lower incidence of TPMT-deficient alleles, with less than 5% of the population having at least one defective allele [60,69,70]. In addition, TPMT polymorphisms are related to only 10% to 25% of the overall thiopurine toxicity [71,72], which limits the value of pretreatment TPMT genotype assessment in Asians [73].
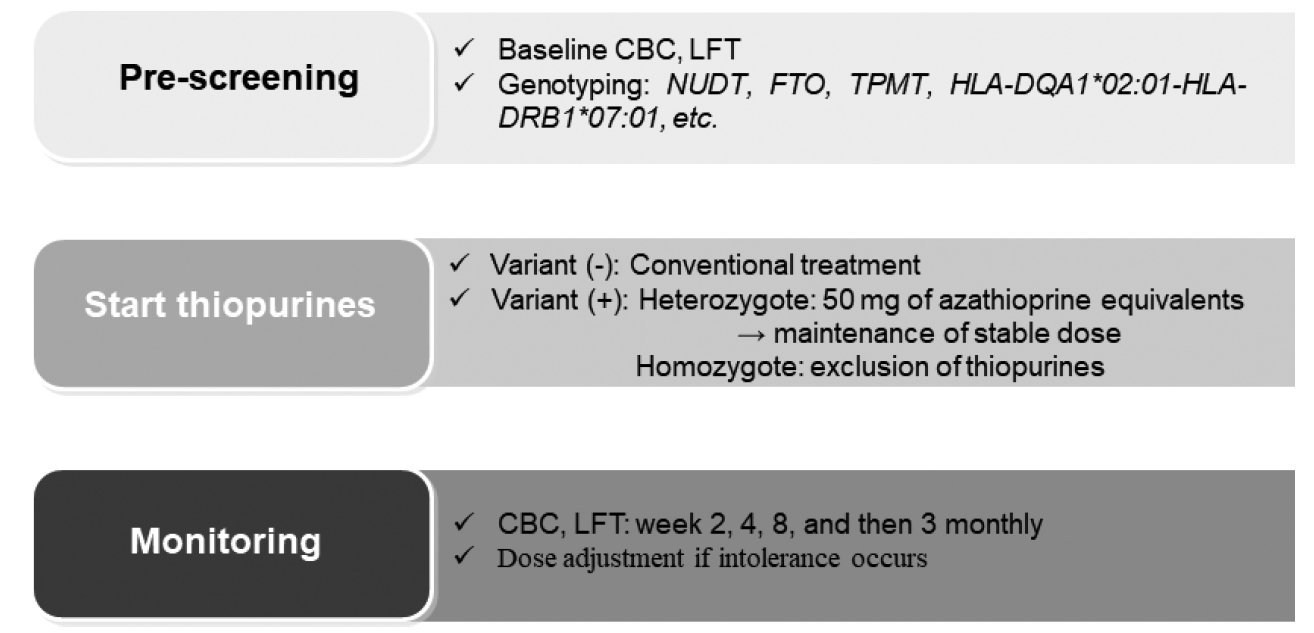
In an effort to find more predictable genetic markers, Yang et al. [69] reported that a missense variant in the nudix hydrolase 15 (NUDT15) (p.Arg139Cys) gene is strongly associated with thiopurine-induced early leukopenia in patients with CD (OR, 35.6; Pcombined=4.88 ×10-94). NUDT15 dephosphorylates the thiopurine active metabolites, thioguanosine triphosphates (TGTP) and deoxy-thioguanosine triphosphate (TdGTP), and prevents the binding of TGTP to Rac1 and the incorporation of deoxy-TGTP into DNA [74]. Thus, the deficiency of NUDT15 increases the levels of thiopurine active metabolites and increases drug toxicity [74]. Compared with the noncarriers of risk alleles, patients with homozygote variants had a lower tolerated dose (0.86 mg/kg/day vs. 1.53 mg/kg/day, P =4.93 ×10-11), shorter interval from onset of therapy to leukopenia (19 days vs. 135 days, P=1.03×10-17), and more severe grade (grade 3 or 4) leukopenia (14 patients vs. 4 patients, P=4.85×10-19) [69].
Compared to TPMT, NUDT15 variants are most frequent in East Asians (9.8%), followed by Hispanics (3.9%), and rarely in Europeans (0.2%) [75]. Thus, the impact of pretreatment NUDT15 tests in preventing early leukopenia is significant, particularly in Asians [54]. The Korean Association for the Study of Intestinal Diseases guidelines recommends pretreatment determination of NUDT15 genotypes, especially in East Asians [52]. In addition, NUDT15 R139C measurement has been approved for clinical use in Japan since 2019 [76].
Recently, a novel coding variant, fat mass and obesity-associated (FTO) p.A134T (rs79206939), using a genome-wide association study was found in IBD patients [77]. FTO belongs to the AlkB family of Fe(II)/α-ketoglutarate-dependent dioxygenases and demethylates a nucleotide [54]. The demethylation function of FTO engages in the abrogation of methyl-TIMP, a thiopurine metabolite, which is a potent purine synthesis inhibitor [78], and the repair of impaired DNA or RNA [79]. Leukopenia occurred in 66.7% of patients with FTO p.A134T and in 32.4% of patients with wild-type FTO. The nucleotide demethylase assay confirmed that the p.A134T variation reduced FTO activity by 65% [77]. Based on this study, a randomized controlled study was conducted to evaluate the value of a tailored therapy with pretreatment genotyping for three genes (TPMT, NUDT15, and FTO) to prevent thiopurine-induced myelosuppression in IBD patients [80]. Pretreatment genotyping of one NUDT15 variant (rs116855232), one FTO variant (rs79206939), and three common TPMT variants (rs1800460, rs1800462, rs1142345) effectively reduced thiopurine-related myelosuppression compared to that in the non-genotyping group (15.4% vs. 42.1%, P= 0.001). Pretreatment genotyping also reduced the number of thiopurine discontinuations or dose reductions during thiopurine administration. Considering the higher frequency of FTO p. A134T in Koreans (5.1%) compared with that in the Western populations (< 0.1%) [77], FTO could be a candidate to predict the risk of thiopurine-induced myelosuppression in East Asian patients [54]. Therefore, further research is necessary.
Besides myelosuppression, pancreatitis is another major thiopurine-induced adverse event affecting 2% to 7% of patients [81,82]. Thiopurine-induced pancreatitis is dose-independent and unpredictable and almost always leads to drug withdrawal [82]. In 2014, a genome-wide association study identified a 2.59-fold increased risk of pancreatitis in IBD patients who had the class II HLA gene region polymorphism (rs2647087 mapped to the HLA-DQA1*02:01-HLA-DRB1*07:01) [83], and which was confirmed in another cohort. The risk of pancreatitis was 0.53% for wild-type (A/A), 4.25% (OR, 4.19; 95% CI, 1.02 to 36.45; P= 0.044) for heterozygous (A/C), and 14.63% (OR, 15.83; 95% CI, 3.80 to 145.26; P= 0.0001) for homozygous variant (C/C) patients [81].
Based on previous studies, the following protocol was proposed for the dosing of thiopurines in IBD patients (Fig. 1). Thiopurine therapy should be personalized based on the individual pharmacogenetic results.
ANTI-TUMOR NECROSIS FACTOR-ALPHA AGENTS
Anti-TNFα agents, mainly infliximab (IFX) and adalimumab, are the most effective treatments for inducing and sustaining clinical and endoscopic remissions in both UC and CD patients [84-88]. Anti-TNFα agents rapidly improve inflammation with onset of effects within 2 weeks [10] and have advanced clinical outcomes including reduction in hospitalizations and surgeries, steroid-free remission, better quality of life, and healing of fistulizing CD [1,89]. Despite their high efficacy, one-third of patients were unresponsive (primary nonresponse), and an additional one-third who initially responded to therapy experienced relapse (loss of response) [90-92]. The formation of anti-drug antibodies, that is, immunogenicity, is an important determinant of therapeutic failure [93,94]. Approximately 65% of patients treated with IFX and 38% of patients treated with adalimumab develop have anti-drug antibodies despite concomitant use of immunosuppressive agents such as thiopurines that reduce immunogenicity [93,95]. Thus, to predict individual responses to anti-TNFα agents, genetic variants involved in immune processes, inflammation, autophagy, and apoptosis have been investigated [10].
In a recent genome-wide study including 1,240 biologic-naïve patients with CD, the HLA-DQA1*05 allele, carried by 40% of Europeans, was identified as a genetic determinant of immunogenicity. Carriership of the HLA-DQA1*05 haplotype increased the immunogenicity regardless of medication (IFX; hazard ratio [HR], 1.92; 95% CI, 1.57 to 2.33) (adalimumab; HR, 1.89; 95% CI, 1.32 to 2.70) and concomitant use of an immunosuppressive agent (anti-TNFα monotherapy [HR, 1.75; 95% CI, 1.37 to 2.22], combination therapy [HR, 2.01; 95% CI, 1.57 to 2.58]) [96]. Another cohort study including 262 IBD patients found that the HLA-DQA1*05 variant increased not only the risk of IFX antibody formation (adjusted HR, 7.29; 95% CI, 2.97 to 17.191; P = 1.46 × 10-5) independent of age, sex, weight, dose, and concomitant use of immunomodulatory, but also the risk of loss of response (adjusted HR, 2.34; 95% CI, 1.41 to 3.88; P= 0.001) and discontinuation (adjusted HR, 2.27; 95% CI, 1.46 to 3.43; P= 2.53× 10-4) [97].
Several candidate genes to predict treatment response based on the pathogenesis of IBD or mechanisms of action of anti-TNFα have also been suggested. In 2014, Bank et al. [98] reported that 19 out of 39 functional polymorphisms in 26 genes that alter the NFκB-mediated inflammatory response (TLR2 [rs3804099, rs11938228, rs1816702, rs469 6480], TLR4 [rs5030728, rs1554973], TLR9 [rs187084, rs352 139], LY96 [MD-2] [rs11465996], CD14 [rs2569190], MAP3K14 [NIK] [rs7222094]), TNFα signaling (TNFA [TNFα] [rs361525], TNFRSF1A [TNFR1] [rs4149570], TNFAIP3 [A20] [rs6927172]), and other cytokines regulated by NFκB (IL1B [rs4848306], IL1RN [rs4251961], IL6 [rs10499563], IL17A [rs2275913], IFNG [rs2430561]) other cytokines regulated by NFκB (IL1B [rs484 8306], IL1RN [rs4251961], IL6 [rs10499563], IL17A [rs227 5913], IFNG [rs2430561]) were associated with response to anti-TNFα therapy in 738 anti-TNFα-naïve IBD patients [98]. In 2019, they replicated and updated the gene signature in a newly established IBD cohort. Ten polymorphisms in genes involved in the regulation of NFκB pathways (TLR2, TLR4, and NFKBIA), TNFα signaling pathway (TNFRSF1A), and cytokine pathways (NLRP3, IL1RN, IL18, and JAK2) were associated with response to anti-TNFα therapy [99].
With extreme phenotype approach that identified patients at opposite ends of IFX response, rs2158962 in TNF receptor associated protein 1 (TRAP1) was found to have a high rate of association with mucosal healing in CD patients (OR, 4.94; Pcombined=1.35×10−7). When the predictive power of rs2158962 was assessed in beyond extreme cases, mucosal healing rate was 5.5-fold higher in homozygotes (P= 6.10× 10−5), and 2-fold higher in heterozygotes (P= 0.024) compared with that in non-carriers. In the dextran sodium sulfate-induced acute colitis mice model, TRAP1 transgenic mice also exhibited a better response in histologic recovery compared with the wild-type mice [12].
Genetic analysis combined with clinical characteristics have been attempted to improve the predictive power of the response to anti-TNFα therapy. Recently, a Korean-German study team identified the clinical and genetic markers involved in IFX response using whole-exome sequencing data. The rs 2228273 in zinc finger protein 133 (ZNF133) (OR, 11.94; P= 2.10×10−5), concurrent azathioprine/6-mercaptopurine use (OR, 4.78; P=0.031), and bodyweight under 50 kg at the first IFX use (OR, 5.26; P = 0.013) were associated with primary non-response. The combined genetic and clinical models had superior predictive power compared to the model including only genetic variables or only clinical variables (area under the receiver operating characteristics [AUROCs], 0.84, 0.70, and 0.69, respectively) [100]. ZNF133 is a Krüppel-associated box-zinc finger family protein [100] and is known to repress S100A4 gene expression [101]. High levels of S100A4, a member of the S100 family of calcium-binding proteins, are associated with a poor clinical IFX response and high occurrence of anti-IFX antibodies in rheumatoid arthritis patients [101].
In a prospective study comprising 231 UC patients, a combined clinical-genetic model and clinical predictive model were compared for primary nonresponse and durable response. For genotyping, a platform with 196,524 polymorphisms (718 small insertion deletions, 195,806 SNPs) of known major immune and inflammatory disease loci was used. With detection of 8 alleles associated with primary nonresponse (potential genes PTPN22, PHTF1, TRAF3IP2-AS1, NFIL3, FIBP, SH2B3, ATXN2, UBAC2, GPR18, IFNGR2, IFNAR1, IL10RB), the combined clinical-genetic model showed higher power than that in the clinical-only model (AUROCs, 0.87 vs. 0.57, P< 0.0001). Interestingly, the identified SNPs involved in primary nonresponse were not associated with IFX antibodies or serum drug levels, which implies that the mechanisms of these genetic loci might be mediated by other than drug pharmacokinetics or antibody formation [102]. Moreover, a Dutch study team recently explored the efficacy of pharmacogenetic passport, including the genetic variation of TPMT, NUDT15, HLA-DQA1*02:01-HLA-DRB1*07:01 (for thiopurine-induced pancreatitis), and HLA-DQA1*05. The pharmacogenetic passport predicted 36% of thiopurine or anti-TNFα adverse responses in 150 cases. The calculated numbers needed to genotype, treat, and harm to prevent one of these adverse drug responses were 24, 9, and 6, respectively. With the estimation of the numbers needed for genotyping, 24 patients should undergo pretreatment genotyping for prevention. This means that if pretreatment genotyping is performed in 24 patients, one adverse drug response would be prevented using an appropriate alternative treatment strategy [103].
CONCLUSION
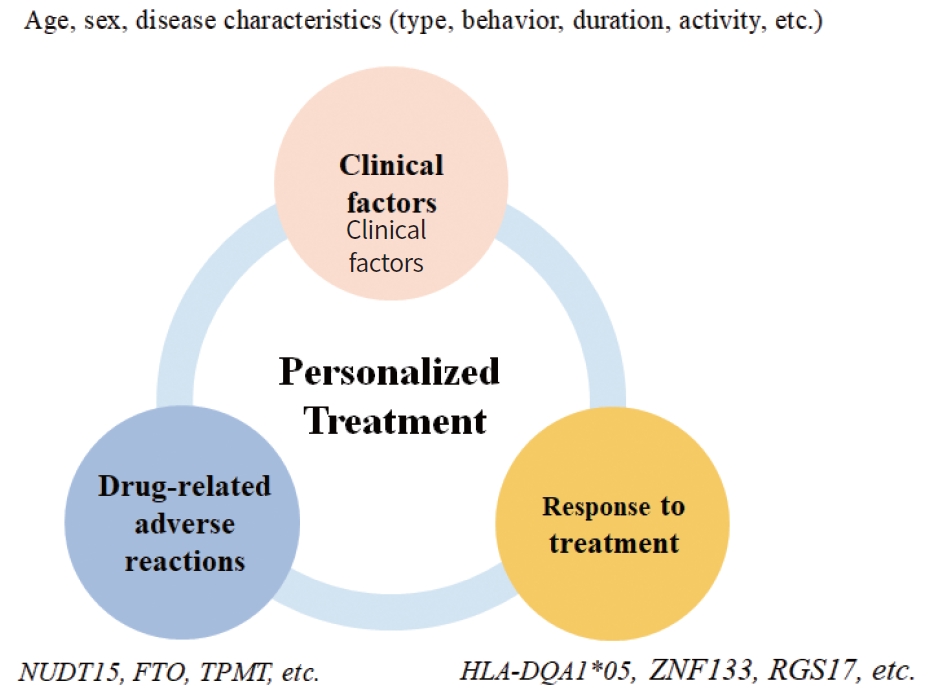
Personalized medicine based on individual pharmacogenetics is necessary to improve clinical outcomes in the management of IBD, which requires long-term treatment. In addition to efficacy and safety issues, personalized therapy would be beneficial in reducing the treatment cost. They could optimize safe and cheap conventional treatment, avoid expensive but ineffective or harmful drugs, especially cost-associated anti-TNFα adverse reactions, and achieve optimal dosing during an early treatment period [9].
However, pharmacogenetics-based IBD treatment in the clinical setting is still challenging, with only several genetic predictive factors for thiopurines being validated and recommended by guidelines (Fig. 2). Studies on other drugs have shown inconsistent results, or include relatively small sample cohorts. To overcome these limitations, future studies involving larger nationwide and international collaborative designs should be conducted and should evaluate combined epigenetic, environmental and clinical features as well as genetic biomarkers [10]. With the development of relevant markers, tools for interpreting genetic information for appropriate clinical implementation could be provided [10].