Biomarkers for gastric cancer: molecular classification revisited
Article information
Abstract
Gastric cancer (GC) is one of the most common lethal cancer worldwide. In recent years, several new targeted therapeutic agents for the treatment of metastatic GC have been developed. These include drugs that block human epidermal growth factor receptor 2 (HER2) or epidermal growth factor receptor, ramucirumab monoclonal antibody that binds to a receptor for vascular endothelial growth factor, and other targeted agents such as sorafenib and apatinib, and immunotherapies. In this short review, we provide a summary of clinical and preclinical biomarkers (HER2, mesenchymal-epithelial transition [MET], fibroblast growth factor receptor 2 [FGFR2], ring finger protein 43 [RNF43], microsatellite instability and mismatch repair, Epstein-Barr virus, programmed cell death ligand-1, and tumor infiltrating lymphocytes) for treatment strategies and will address the molecular classification of GC revisited with an aim to select the best-precision treatment strategies for GC patients.
INTRODUCTION
The American Cancer Society’s estimates for stomach cancer in the United States for 2017 are about 28,000 cases of gastric cancer (GC) will be diagnosed (17,750 in men and 10,250 in women) and about 10,960 people will die from this cancer (6,720 men and 4,240 women) (https://www.cancer.org). Stomach cancer is much more common in other parts of the world including Korea, China, and Japan. In patients diagnosed with advanced GC disease stage, the patients have a dismal prognosis with a high mortality rate.
Chemotherapy target cells that divide rapidly; however, there are other aspects of cancer cells that make them different from normal cells and many researchers have developed new drugs targeting those differences. As a result of the rapid advancements in the field of tumor biology, attention has been focused on the new modality of molecular targeted therapy for advanced cancer [1]. Therefore, targeted drugs may work in some cases when standard chemotherapy does not work with fewer side effects than standard chemotherapy.
Molecular targeted inhibitors effectively regulate overexpressed molecules in tumor cells and the signaling pathways that are closely associated with tumorigenesis; thereby, modulating the biological behavior of tumor cells [2]. The genomic landscape of GC is highly heterogeneous [3]. Therefore, it is difficult to target the entire tumor because subclones of GC cells exhibit different biological behaviors. To date, only two targeted treatments, trastuzumab, and ramucirumab, have become part of the treatment paradigm for GC, partly due to difficulties in defining predictive biomarkers in this disease [4]. There are many promising drugs in the pipeline and this article will describe the targets (biomarkers) associated with GC treatment and other potential novel approaches targeting DNA repair deficiencies and the immune system and will include human epidermal growth factor receptor 2 (HER2), mesenchymal-epithelial transition (MET), fibroblast growth factor receptor 2 (FGFR2), ring finger protein 43 (RNF43), microsatellite instability (MSI), mismatch repair (MMR), Epstein-Barr virus (EBV), programmed cell death ligand-1 (PD-L1), and tumor infiltrating lymphocytes (TILs).
HER2
HER2 immunohistochemistry in gastric cancer: what is different from the breast HER2?
HER2 protein overexpression and gene amplification are more heterogeneous in GC and require a different immunohistochemistry (IHC) scoring system compared to breast cancer (HER2 testing in GC: recommendations of an Asia-Pacific Task Force). Heterogeneous staining is a true biological feature that is far more common in gastric and gastroesophageal junction carcinomas than in breast carcinomas [5] and is estimated to be present in up to 30% of HER2-positive GC cases. Heterogeneous HER2 overexpression was observed in 33% of HER2-positive GC cases using quadruplicated tissue microarray study. Discrepancy was also observed in 20% of paired primary and distant metastases in HER2-positive GC cases [6]. These results suggest that intratumoral heterogeneity and discrepant HER2 results in primary and metastatic tumor are not uncommon in GC. Heterogeneity is more often detected in IHC 2+ cases or in mixed histologic type by Lauren often associated with chromosome 17 polysomy (≥3 CEP17 [chromosome enumeration probe 17] signals per nuclei) (Fig. 1) [7].
HER2 (human epidermal growth factor receptor 2) 2+ with chromosome 17 polysomy. (A) Immunohistochemistry finding showing 2+ stained in the membrane of tumor cells. (B) Silver in situ hybridization with chromosome 17 polysomy (×40).
Factors affecting gastric HER2 positivity and how can we overcome
The overall frequency of HER2 overexpression in GC ranged from 7% to 53.4% with a mean of 17.9% [6]. This wide range of overexpression of HER2 might be explained by location within the stomach (higher in upper third compared to lower stomach), histologic subtype (34% in intestinal subtype compared to 6% in diffuse-type), disease stages, interpretation of IHC results, antibodies used for IHC [8] and storage duration of paraffin blocks. For proper HER2 IHC testing in gastric biopsy samples, it is recommended to have at least four pinch biopsy tissue fragments containing cancer cells [9] and surgical specimens from patients that previously obtained HER2-negative results in biopsies should also be tested to increase the chance of finding HER2-positive tumors [10]. To overcome factors affecting gastric HER2 positivity, we should aware the importance of quality assurance and quality control of HER2 testing including careful interpretation, use tissues with more carcinoma cells for biopsy specimen (≥4 cancer tissue fragments), selection of intestinal histology in operation specimen, and test both biopsy/surgical specimens and both primary/metastatic specimens.
Genomic alterations associated with HER2-positive GC
In patients with HER2-positive GC, tumor protein p53 (TP53) mutation is the most frequent genetic alterations followed by cyclin-dependent kinase Inhibitor 2A (CDKN2A, 4%), and phosphatidylinositol-4,5-bisphosphate 3-kinase, catalytic subunit α (PIK3CA) and Kirsten rat sarcoma viral oncogene homolong (KRAS) mutations (2%) [11], and five genes were concomitantly co-amplified: G1/S-specific cyclin-E1 (CCNE1, 8%), PIK3CA (8%), KRAS (2%), cyclin-dependent kinase 4 (CDK4, 2%), and CDK6 (2%). These results are consistent with our previous molecular classification study using 300 GC samples, TP53 pathway inactive subgroup was characterized by TP53 loss through deleterious mutations in TP53 or MDM2 (mouse double minute 2 homolog) amplification and characterized by both focal amplifications in oncogenes such as HER2 (17.4%), epidermal growth factor receptor (EGFR, 7.0%), MET (3.5%), and CCNE1 (17.5%) as well as large scale chromosomal gains and losses. However, HER2-positive GC with concomitant MET and/or EGFR overexpression can be demonstrated in small subsets of GC associated with aggressive behavior [12]. Moreover, all patients with concomitant CCNE1 amplification and HER2 amplification progressed shortly after trastuzumab-based chemotherapy [11]. Hence, in this subset of patients, combination therapy should be tested in the context of clinical trials.
MET (MET PROTO-ONCOGENE, RECEPTOR TYROSINE KINASE)
How to select MET-amplified GC using IHC: a new interpretation method
The establishment of selection criteria in identifying subpopulations that may benefit from treatment is a key aspect of further development of anti-MET monoclonal antibody (METMab) and hepatocyte growth factor/scatter factor monoclonal antibody treatment [13]. The study with MetMAb in non-small cell lung cancer and the study with AMG102 in GC highlight an essential requirement for patient stratification to ensure clinical benefit: high MET expression in cancer cells [14]. In that study, MET overexpression assessed by new interpretation method based on membranous and cytoplasmic staining correlated well with increased copy number gain, mRNA levels and MET gene amplification, and was an independent prognosticator of poor survival, supporting the clinical impact of MET proto-oncogene activation in GC.
MET overexpression caused by MET exon 14 deletion
The discordance between low MET amplification and high MET protein expression indicates there are other potential mechanisms that can lead to MET overexpression. MET exon 14 deletion (METex14del) has been postulated to be one potential mechanism for MET protein overexpression (Fig. 2) [15]. In our previous screening with nCounter fusion transcript analysis with mRNA, we found three GC cases with poorly differentiated adenocarcinoma, METex14 and MET IHC 3+ in both membranous and cytoplasmic staining, in which concomitant MET gene amplification was observed in one case (a 27-year-old male patient with massive malignant ascites and died shortly after diagnosis). His tumor showed strong MET overexpression by IHC (3+) without MET amplification. A selective small molecule inhibitor of Met (PHA-665752) and cabozantinib demonstrated potent growth inhibition in METex14del+ patient-derived GC tumor cell line [15].
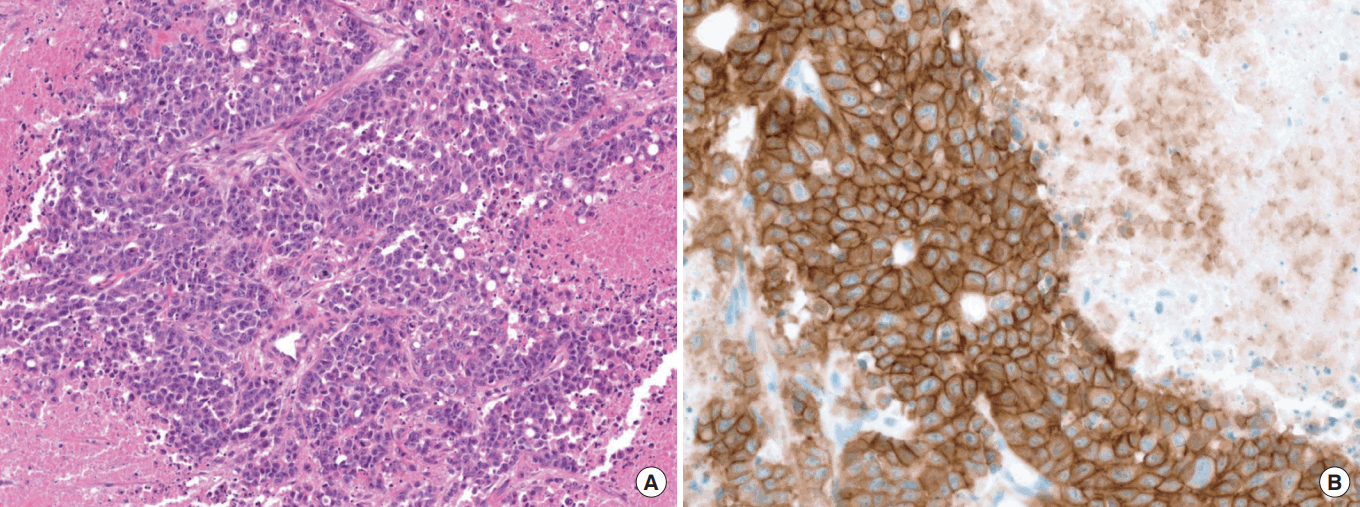
Mesenchymal-epithelial transition (MET) immunohistochemistry in a gastric cancer case with METex14del (MET exon 14 deletion). H&E photomicrograph of gastric carcinoma (A) with MET immunohistochemistry showing both membranous and cytoplasmic strong (3+) staining (B) (×40).
FGFR2
FGFR2 in gastric cancer: how can we detect?
FGFR2 gene amplification, and resulting FGFR2 protein overexpression, is rare in GC patients, and development of an accurate and widely available method for mass screening to identify patients who may respond to treatment with FGFR inhibitors is important. Recently, we found FGFR2 IHC results correlated very well with those of copy number variation [16]. Interestingly, FGFR2 amplification by fluorescence in situ hybridization was observed in 100% of the IHC ≥1+ cases. Given that several FGFR inhibitors are now in under clinical trials for treatment of metastatic or unresectable GC harboring FGFR2 amplification or FGFR2b overexpression, IHC can be a powerful tool for massive screening of this small subset of GC patients.
Clinical significance of FGFR2
FGFR2 overexpression is more frequently observed in metastatic lymph nodes compared to matched primary GC. Intriguingly, analysis of paired primary and metastatic GC tumors revealed more FGFR2-positive tumor cells in metastases than in primary GC. We also observed frequent FGFR2 overexpression within lymphatic tumor emboli, which was noteworthy (Fig. 3). Based on marked intratumoral heterogeneity, frequent focal overexpression/amplification of FGFR2 within lymphatic tumor emboli, and preclinical studies, FGFR2 amplification may play important roles in tumor progression, particularly in lymphangitic metastasis of GC, and the potential therapeutic effect from targeted therapy can be expected in patients with this GC subset [16].
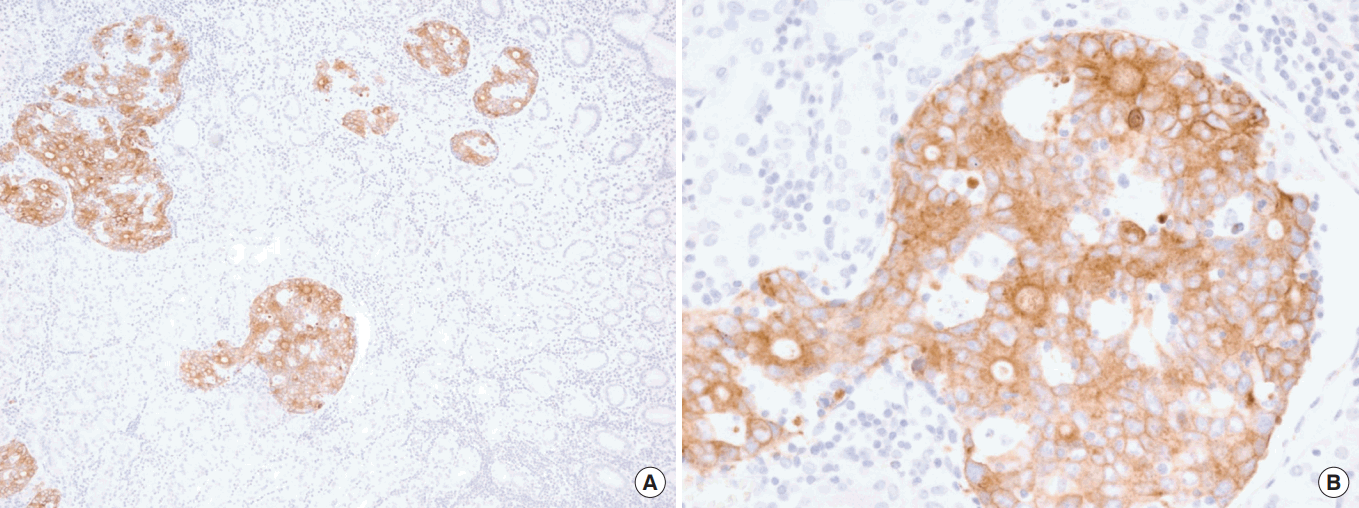
FGFR2 (fibroblast growth factor receptor 2) 3+ stained within lymphatic tumor emboli (A) with lower magnification (×10) and (B) higher magnification showing carcinoma within lymphatic spaces (×40).
RNF43
Clinical significance of RNF43
Ubiquitin E3 ligase RNF43 is a transmembrane E3 ubiquitin ligase that potently inhibits Wnt signaling through targeting Wnt receptor for degradation and has been suggested as a negative regulator of Wnt signaling and found to be frequently mutated in many cancers including GC [17,18]. Several agents, including porcupine inhibitor, frizzled antibody, and LRP6 (low-density lipoprotein receptor-related protein 6) antibody, are being developed to inhibit ligand-induced Wnt/β-catenin signaling. Previous preclinical observation that all pancreatic cancer cell lines showing strong Wnt dependency carry RNF43 mutations (homozygous mutations of RNF43, F69C, and M18fs) suggests that the presence of RNF43 mutations may serve as a predictive biomarker for selecting patients likely to respond to upstream Wnt pathway inhibitors [19]. However, as the preclinical studies were performed in pancreatic cancers, further studies in GC are needed to validate this conjecture [17].
Prevalence of RNF43 mutations in GC
Previous two studies on GC reported that mutation rate of RNF43 was higher in MSI subtype than that of microsatellite-stable (MSS) subtype [20,21]. Precisely, RNF43 was mutated in 4.8% of MSS and 54.6% of MSI tumors. Among them, 62.5% of mutations were truncating, and only four MSS GC samples (4.4%) harbored missense mutations of RNF43. In The Cancer Genome Atlas (TCGA) data, RNF43 was mutated in six out of 215 MSS GC samples, consisting four frameshift and two nonsense mutations. Unexpectedly, four RNF43 mutations were from genomically stable subtype [21]. These observations suggest that truncating mutations of RNF43 in MSS GC would be a good candidate biomarker for therapeutic agents specifically targeting the Wnt/β-catenin signaling.
MSI AND MMR DEFICIENCY
Clinical significance of MSI and MMR
TCGA and Samsung Medical Center-Asian Cancer Research Group (SMC-ACRG) GC studies proposed a molecular classification dividing GC into four subtypes and MSI-GC is one of them. MSI is associated with elevated mutation rates, including mutations of genes encoding targetable oncogenic signaling proteins [21]. The incidence of MSI GC was previously reported to be 8.5% to 37.8% [22]. MSI-GC is exclusive with EBV-positive GC and more frequent in antral locations (distal stomach), intestinal type be Lauren, female, older ages, and earlier disease stages than tumors within body/cardia, diffuse histology, male, younger patients, and advanced stages [23,24].
Prognosis of MSI-GC
The prognostic significance of MSI in GC has been controversial [25]. In the previous largest cohort study with consecutive 1,990 GC patients who had undergone curative gastrectomy, the disease-free survival curves showed no significant differences between MSS and MSI patients at each stage of I, II, III, and IV. However, given the detection of MSI in lower disease stages, direct comparison with all MSS-GC would result in better survival [23,26-28]. This lower disease stage and better prognosis would be related with increased TIL found in this subtype. However, as MSI is also detected even in a higher disease stage, in that case, the prognostic significance disappears. Overall, the prognostic significance of MSI would depend on disease stage or sample cohort characteristics rather than MSI itself.
Underlying MMR deficiencies in MSI-GC
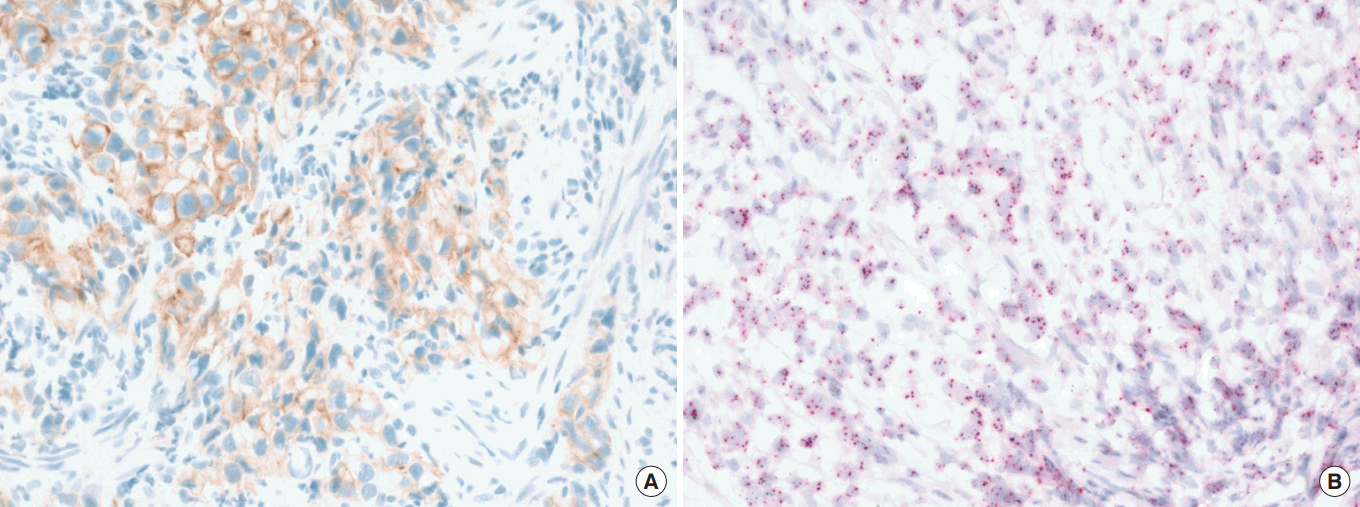
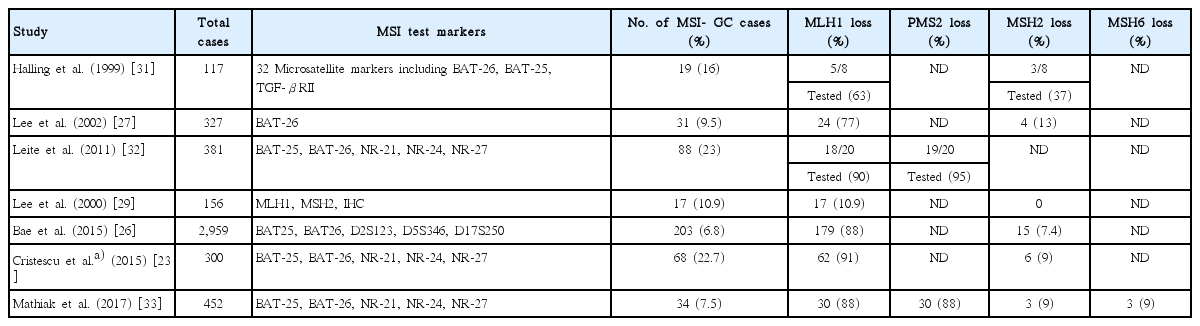
In MSI-GC, in >90% of cases are associated with MutL homolog 1 (MLH1) and/or PMS2 losses and are associated with hypermethylation of hMLH1 gene. This type of MSI occurs early during gastric multistep carcinogenesis through adenoma-carcinoma found in 30% of cases [29,30]. These observations explain the frequent occurrence of frequent MSI in older patients with distal gastric location, intestinal histology and earlier disease stage, and finally a favorable prognosis. In less than 10% of cases with MSI-GC, MutS protein homolog 2 (MSH2), and/or MSH6 losses are found. However, depending on selection of markers for MSI, the frequencies varied from 0% to 37% (Table 1) [23,26,27,31-33]. Given the shortcomings of missing standardized testing algorithms with prevalence of MSI-GC ranging from 0% to 44.5% [33] and highly variable MSH2 expression losses, future comprehensive diagnostic algorithm of MSI tests are needed in GC and this is urgently needed because MSI is an important biomarker for immunotherapy, one of the most exciting and new discoveries in recent cancer therapy.
MMR protein alterations in sporadic MSI-GC cases
Importance of MSI in a new era of immunotherapy
Responses to immune checkpoint inhibitors have been observed in tumors with both low and high PD-L1 expression. A strong putative biomarker has emerged in MSI-high status, which results either from germline mutation of DNA MMR genes, as seen in patients with Lynch syndrome, or in tumors with promoter hypermethylation leading to potential gene silencing of DNA MMR pathways.
MSI status results in a higher tumor mutational burden, and patients with MSI colorectal and noncolorectal cancers have shown striking responses to anti–PD-1 therapy in recent trials of pembrolizumab [34]. Whether or not the same is observed in MSI esophagogastric cancers remains to be established (http://www.ascopost.com/issues/june-25-2016/anti-pd-1-treatment-with-pembrolizumab-in-gastricgastroesophageal-junction-cancers-who-is-likely-to-respond). MSI GCs showed signifi cantly higher rates of PD-L1 expression compared with MSS GCs [35-37]. Genomic profiling has also identified the MSI-GC subtype shows higher expression of the ligands PD-L1 and PD-L2 [21]. Recently we observed frequent overexpression of PD-L1 in MSI-GC in 61.5% of cases and unexpectedly, we found that PD-L1 expression is independently associated with longer survival even within MSI-GC cases [25].
EBV
Clinical significance of EBV+GC
EBV-positive GC (EBV+GC) is one of the four subtypes of GC, as defined by TCGA having several clinicopathological features such as longer survival and higher frequency of lymphoepithelioma-like carcinoma (LELC) or increased TIL (also known as Crohn-like reaction [CLR]) that distinguish it from EBV-negative GC (EBVnGC). TIL constitute the principal cellular component of the immune-active tumor microenvironment and their presence contributes to improved survival of patients with cancer [38]. The number of TILs increases in EBV+GC, especially in LELC or CLR. The presence of EBV+ tumors negatively correlates with the TNM (tumour, node, and metastasis) stage parameter as well as with the values of its individual components (primary tumor site, regional lymph node involvement, or presence of distant metastasis) [39]. Overall, EBV positivity is associated with favorable prognosis and this survival difference is mainly achieved by a degree of TIL within the microenvironment of cancer [40].
Deregulated immune response and TIL
Recently, we reported a gene expression profile analysis to compare tumor and non-tumor gastric tissues from 12 patients with EBV+GC and found that EBV+GC had a higher degree of genetic homogeneity than EBVnGC cases. Deregulation of gene expression affected fewer genes in EBV+GC compared to EBVnGC with significantly altered expression signals especially in cytokine (chemokine) pathways [41]. These changes could recruit reactive immune cells to the intra- or peritumoral area of EBV+GC (can be designated as TIL) and this circumstance might contribute to longer survival of patients with EBV+GC compared to EBVnGC and make EBV+GC as a distinct tumor with prominent inflammatory infiltrates. EBV+GC has evidence of hyperactive adaptive and innate immunity, with evidence of T-cell activation via the cytokines interleukin 2 (IL-2), IL-12, IL-23, and IL-27 [21,42].
Biomarkers in EBV+GC
Several studies found that EBV+GC is often accompanied by more extensive infiltration of CD8-positive cytotoxic T cells, FoxP3+ T regulatory T cells and higher interferon-γ expression, which induce the expression of indoleamine 2,3-dioxygenase (IDO1), a potent immune cell inhibitor, in EBV+GC cells [25,43]. Recent genomic profiling studies revealed frequent somatic mutations of PIK3CA (10.3% to 80%), AT-rich interactive domain-containing protein 1A (ARID1A, 47% to 55%), v-akt murine thymoma viral oncogene homolog 2 (AKT2, 38.2%), and B cell lymphoma 6 corepressor (BCOR, 23%) genes [44] and amplification of Janus kinase 2 and PD-L1/L2 [21]. Recent IHC study in 32 EBV+GC observed overwhelming PD-L1 staining in tumor cells in 50% and immune cells in 94% of cases. Moreover, interferon-γ driven gene signature, an additional proposed marker of sensitivity to PD-1 therapy, were also enriched in EBV+GC suggesting that patients with EBV+GC may have greater likelihood of response to PD-1 blockade [45]. Therefore, in EBV+GC patients, the combination of anti–PD-1/PD-L1 agents and IDO inhibitor would be active areas of further investigation in cases failed to conventional chemotherapy or PIK3CA/AKT/mTOR inhibitor therapies.
PD-L1 AND TIL
After years of controversy, it is now recognized that the immune system can play a role in the control of tumor growth and progression, a process known as cancer immunoediting [46]. The host immune system can also contributes to the efficacy of some cancer therapies where the tumor death induced may be “immunogenic” [47]. Recently, classification of tumors into four groups on the basis of their PD-L1 status and presence or absence of TILs has already been proposed [46,48]. These include type I (PD-L1+ with TILs driving adaptive immune resistance), type II (PD-L1 negative with no TIL indicating immune ignorance), type III (PD-L1+ with no TIL indicating intrinsic induction), and type IV (PD-L1 negative with TIL indicating the role of other suppressors in promoting immune tolerance). The proportions of various human tumors that fit into each of these types, as defined by TILs/PD-L1 status, likely depends on the genetic aberrations and oncogene drivers of the cancer as well as the tissue they arise in [46]. Although this classifications were well described and validated in melanoma, analogous frequencies in GC generated by the same methodologies are not available. Future comprehensive studies are strongly required in GC. Tailoring cancer immunotherapy based on type of tumor microenvironment is well described in a recent review paper [46].
CONCLUSION
We reviewed the present and emerging biomarkers in GC. There are efforts to classify GC based on molecular findings with clinical significances [21,23]. Based on previous classifications and biomarkers, we revisited new molecular classifications of GC for precision cancer therapy (Table 2). In a new era of immunotherapy of GC, we urgently need an accurate and sensitive method to detect MSI in GC and classification of GC into four groups by PD-L1 and TIL status to explore mechanisms underlying response or resistance for immunotherapy.

Four gastric cancer subtypes based on clinical findings and genomic alterations
Notes
No potential conflict of interestrelevantto this article was reported.
Acknowledgements
This work was supported by a grant from the Korean Health Technology R&D Project, Ministry of Health and Welfare, Republic of Korea (HI14C3418, HI16C1990). Support was also provided by a grant from the 20by 20 project of Samsung Medical Center(GF01140111).