INTRODUCTION
Monogenic diabetes is commonly caused by single-gene mutations with more than 50 causative genes identified. This disease ranges from 1% to 5% in all cases of diabetes and is less affected by behavior and environment [1,2]. Neonatal diabetes mellitus (NDM) and maturity-onset diabetes of the young (MODY) account for a major proportion of monogenic diabetes, while syndromic diabetes constitutes a smaller proportion [3,4]. Over the past decades, diagnosis of monogenic diabetes has improved from being based on clinical phenotypes to molecular genetics, with significant advancement of genome sequencing skills.
Precision medicine is based on tailoring treatment for evidence-based individualization [5]. Challenges faced when implementing precision medicine in diabetes are disease heterogeneity, various genetic influences, difficulties of accurate diagnosis, limitations of current treatment, and social, environmental, and psychological factors [6]. This review will explain how successfully precision medicine is implemented in monogenic diabetes by examining the distinct etiology and subgroups that contribute to predicting and treating clinical phenotypes associated with the disease. Furthermore, we will also review the recent Korean studies and suggest methods of prioritizing patient screening for genetic testing.
NEONATAL DIABETES
NDM is defined as developing within the first 6 months of life. Before the availability of genetic screening, NDM was classified based on the clinical course of transient NDM (TNDM), permanent NDM (PNDM), or a specific syndrome. The development of targeted sequencing has allowed for rapid diagnosis of all known genetic etiologies and subsequent precision treatment for NDM [7-10].
Clinical characteristics of TNDM
Patients with TNDM by 6q24 methylation are characterized by hyperglycemia during the first week of life, which resolves by age 18 months [7]. Many of them require exogenous insulin treatment at diagnosis and during remission, while some of them are well responsive to sulfonylurea or a combination of sulfonylurea and inulin. However, approximately 50% of patients relapse during late childhood or early adulthood; therefore, an accurate record of the patient’s history of TNDM is essential. There is no guideline for treatment for relapse, some patients can be treated with sulfonylurea or a combination of sulfonylurea and insulin [11,12]. Commonly, TNDM is caused by overexpression of imprinted genes on chromosome 6q24 (pleomorphic adenoma gene 1 [PLAG1] and hydatidiform mole associated and imprinted [HYMAI]) due to paternally inherited duplications, paternal uniparental disomy, or maternal hypomethylation. PLAG1 and HYMAI encode a zinc finger protein (ZEP) and long noncoding RNA, respectively. Notably, patients with mutations on 6q24 have low birthweight and early development compared to patients with potassium adenosine triphosphate (KATP) channel mutations [13]. The potassium inwardly rectifying channel subfamily J member 11 (KCNJ11) gene encoding the Kir6.2 subunits of the KATP channel and ATP binding cassette subfamily C member 8 (ABCC8) encoding the sulfonylurea receptor 1 (SUR1) subunit of the KATP channel, whose mutations induce PNDM more frequently, are the remaining causes of TNDM.
Clinical characteristics of PNDM
The most frequent causes of PNDM are heterozygous gain-of function mutations in KCNJ11 and ABCC8, while homozygous or some heterozygous loss-of-function mutations in KCNJ11 and ABCC8 develop congenital hyperinsulinism [9,14]. These KATP mutations impede KATP channel closure in multiple ways in both TNDM and PNDM [15]. Additionally, mutations in ABCC8 are more prevalent in TNDM, while mutations in KCNJ11 are more prevalent in PNDM. In TNDM and PNDM, KATP-related diabetes responds well to high-dose sulfonylureas rather than insulin.
PNDM is associated with specific syndromes. Immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome is caused by forkhead box P3 (FOXP3), which is an autoimmune disease characterized by persistent diarrhea, hemolytic anemia, exfoliative dermatitis, autoimmune thyroiditis, and PNDM. Wolcott-Rallison syndrome is an inherited autosomal recessive disease caused by a mutation of eukaryotic translation initiation factor 2-alpha kinase 3 (EIF2AK3), which leads to pancreatic β-cell apoptosis, as well as other clinical characteristics, including mental retardation, renal failure, and early mortality [16]. Approximately 80% of patients with PNDM have known genetic etiology; hence, whole exome sequencing remains a continuous effort [17].
MODY AND PRECISION TREATMENT OF SUBTYPES
MODY is the most prevalent form of monogenic diabetes and was first identified in 1974 by Fajans and Bell [3] and Tattersall [18]. The prevalence of MODY is estimated to be at least 1.2% according to a population-based study conducted in the United States [1]. Currently, 14 genes are identified as disease-causing mutations in MODY patients (Table 1) [19,20]. According to the most comprehensive study that was conducted in the United Kingdom, hepatocyte nuclear factor 1 α (HNF1A, 52%) and glucokinase (GCK, 32%) are the most common causes of MODY, followed by HNF4α (10%) and HNF1β (6%) [21]. It is thought that the study methodology, such as body mass index (BMI), cut-off age, and genetic testing method, influence the prevalence and subtype of MODY between ethnic groups and conducted studies.
GCK-MODY
GCK-MODY is caused by heterozygous inactivating mutations in the glucose sensor enzyme GCK [22]. GCK stimulates glucose metabolism and promotes insulin secretion through multiple mechanisms, including protein stability, enzymatic activity, and increased interaction with the GCK regulator. Less heterogeneity is observed in patients with GCK mutations, who have mild, lifelong, asymptomatic fasting hyperglycemia in the pre-diabetes range. Some patients exhibit diabetes due to type 2 mixed with environmental and hereditary factors. Hyperglycemia in GCK-MODY is less responsive to insulin and oral agents and does not appear to be associated with micro- or macrovascular complications; therefore, these patients seldom need glucose-lowering treatment, except during pregnancy [23]. Maternal GCK mutations can result in macrosomia due to increased insulin secretion in response to hyperglycemic uterine conditions. However, insulin treatment during pregnancy increases the risk of low gestational age in a fetus with GCK mutations; due to the decreased insulin secretion and glucose level below the fetal set point [24]. Therefore, it is ideal to know whether a fetus has mutations. Recently, droplet digital Polymerase chain reaction (PCR) has been used to determine fetal mutations noninvasively by extracting cell-free DNA from a maternal blood sample [25].
HNF1A-MODY and HNF4A-MODY
HNF1A-MODY encodes homeodomain-containing transcription factors related to β-cell development and insulin secretion and is the most prevalent form of monogenic diabetes in the United Kingdom [21]. Over 400 variants of HNF1A have been identified [26]. HNF1A is commonly diagnosed at an earlier age and with a lower BMI than type 2 diabetes, although the risk of long-term micro- and macrovascular complications is similar [27]. HNF1A regulates the expression of sodium-glucose cotransporter 2 (SGLT2), resulting in glycosuria without definite hyperglycemia [28]. Therefore, early glycosuria can serve as a diagnostic indicator for patients with HNF1A mutations, and caution is required when treating these patients with SGLT2 inhibitors [29].
HNF4A-MODY is caused by mutations in genes that encode hepatocyte nuclear factor 4α. These variants are less common than HNF1A mutations but have similar clinical features and treatments. Patients with HNF1A or HNF4A variants have an excellent response to sulfonylurea, which binds to the potassium channel and depolarize the β-cell, resulting in insulin secretion [30]. The standard sulfonylurea dose used to treat type 2 diabetes can cause hypoglycemia in these patients; therefore, a lower dose is recommended. Despite sharing similar characteristics, neonates with HNF4A mutations show increased insulin secretion, leading to macrosomia and transitory hyperinsulinemic hypoglycemia. However, after neonatal hyperinsulinemia occurs, insulin production decreases, and diabetes develops; this cause of diabetes remains unclear [31]. A glucagon-like peptide 1 receptor agonist or dipeptidyl peptidase-4 inhibitor with sulfonylurea was effective without occurring hypoglycemia in HNF1A-MODY, suggesting that HNF1A or HNF4A-MODY patients may benefit from treatment [32,33].
HNF1B-MODY
HNF1B mutations are highly uncommon, accounting for fewer than 1% of MODY patients. HNF1B variants cause MODY type 5 and are associated with multiorgan disease, including renal abnormalities (predominantly renal cysts), abnormal liver function tests, and neurocognitive defects. Approximately 50% of these variants are due to mutations in the HNF1B gene, while the remainder is due to a 17q12 deletion spanning 15 genes, including HNF1B [34]. Although some patients initially respond to sulfonylureas or repaglinide, pancreatic hypoplasia and hepatic insulin resistance eventually necessitate insulin therapy [35]. Therefore, not only diabetic specialists but also nephrologists, urologists, and gynecologists should manage HNF1B-MODY.
SYNDROMIC DIABETES
Monogenic diabetes can develop multisystem syndromes. Diabetes can be a first manifestation of multisystem syndromes.
Wolfram syndrome
Wolfram syndrome is a rare disease caused by recessive mutations in the wolframin ER transmembrane glycoprotein (WFS1) [36]. This syndrome is characterized by diabetes insipidus, optic atrophy, insulin-dependent diabetes, and sensorineural deafness. Diabetes is usually diagnosed in the first decade of life, and accurate and prompt diagnosis is necessary due to the high morbidity and mortality rates. Recently, some WFS1 mutations have been reported to cause isolated diabetes with reduced penetrance for other wolfram syndrome related features [37].
Mitochondrial diabetes
Mitochondrial diabetes is also known as maternally inherited diabetes and deafness. This type of diabetes is caused by a pathogenic mutation in the mitochondrial DNA, usually m.3243A>G [38]. Diabetes is typically diagnosed in the third or fourth decades of life. The hearing loss is usually diagnosed before diabetes, and is bilateral, sensorineural, and progressive [39]. Other rare forms of mitochondrial diabetes comprise of cardiomyopathy, myopathy, nephropathy, and central neurological features in a rare form of mitochondrial diabetes. The penetrance is estimated to be approximately 100% by the age of 70 years. These patients have impaired insulin secretion; therefore, insulin treatment is necessary for most patients.
Lipodystrophy
Monogenic lipodystrophy is a heterogeneous group, characterized by a complete or partial lack of adipose tissue and adipose tissue-derived hormones. This develops insulin resistance and metabolic complications. Due to the lack of adipose tissue, dyslipidemia results in insulin resistance by spillover of fat into ectopic areas [40]. These patients can be classified into congenital generalized lipodystrophy (CGL) and familial partial lipodystrophy (FPLD). CGL is an autosomal recessive disorder and is usually caused by mutations in 1-acylglycerol-3-phosphate O-acyltransferase 2 and Berardinelli-Seip congenital lipodystrophy 2 [41]. These patients have common clinical features such as diabetes, generalized lipodystrophy, and dyslipidemia. On the other hand, FPLD is typically caused by lamin A/C or peroxisome proliferator-activated receptor γ. These patients have body fat deficiency on the buttocks, hips, and limbs.
PRECISE APPROACHES TO MONOGENIC DIABETES
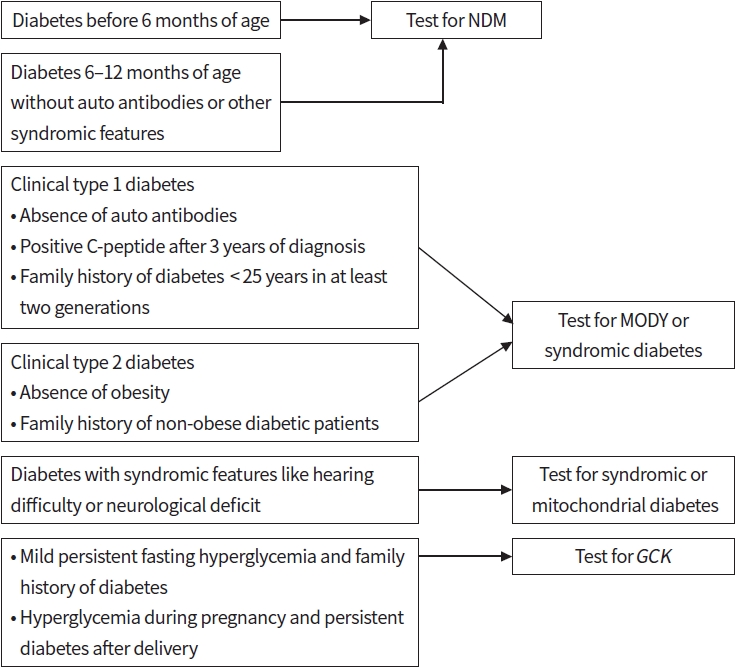
The diagnosis of monogenic diabetes has increased but is still gradual, although genetic testing is quite common even during pregnancy [25,42]. Based on genetic testing, precise treatment for monogenic diabetes has become a novel concept for diabetologists. The International Society for Pediatric and Adolescent Diabetes recommends genetic testing for diabetes patients younger than 6 months and without autoantibodies if younger than 12 months [43]. Moreover, it helps the clinician predict prognosis and improve care, including for patients with KATP mutations treated with oral sulfonylurea [10]. The genetic test is cost-effective for these young patients. However, evaluating young adults and children with type 1 and type 2 diabetes presents certain difficulties, although onset age, endogenous insulin secretion, absence of obesity, and absence of autoantibodies help distinguish MODY from type 1 and type 2 diabetes [21,44,45]. Consequently, there is no single criteria that may accurately identify MODY patients. Using the ideal cut-offs, Shield et al. [46] developed the ‘MODY Probability Calculator,’ which may be accessed for free at www.Diabetesgenes.org. Additionally, incorporating MODY characteristics into this model can result in an improved diagnosis of MODY.
GENETIC TESTING AND INTERPRE TATIONS
Using a next-generation sequencing (NGS) panel, a patient’s entire exome or all MODY genes can be examined simultaneously. Sanger confirmation may be required after variants are found in NGS; however, exome analysis can miss mutations in noncoding regions, deep intronic regions, and 5’- and 3’-untranslated regions (UTRs) associated with monogenic diabetes [47]. Using exome or genome sequencing for monogenic diabetes frequently identifies novel variants; therefore, caution must be exercised when differentiating disease-causing mutations from benign variants. The American College of Medical Genetics and Genomics and the Association for Molecular Pathology published guidelines for interpreting sequence variants in 2015 [48]. According to these guidelines, variants must be classified as pathogenic, likely pathogenic, of uncertain significance, likely benign, and benign. After the first publication of guidelines, revised recommendations regarding loss-of-function and uses of functional evidence were provided [49,50].
OTHER BIOMARKERS FOR MODY
High-sensitivity C-reactive protein (hs-CRP) is emerging as a novel biomarker for MODY [51]. Moreover, an HNF1α binding site in the CRP gene is involved in regulating the basal constitutive CRP in the liver [52]. Patients with HNF1A variants have significantly lower hs-CRP than others, including type 1 diabetes, type 2 diabetes, and GCK-MODY [53,54]. However, the Singapore study did not have a better diagnostic yield based on the hs-CRP criteria. It is assumed that various factors, such as BMI and race, affect the hs-CRP level [55]. Therefore, the appropriate cut-off value for hs-CRP is needed, and caution is required for its efficacy in the Asian population in particular. Additionally, plasma fucosylated glycan and apolipoprotein M are promising candidates [56,57]. Recently, microRNA (miRNAs) has been used as a promising biomarker; Bonner et al. [58] reported the use of circulating miRNAs in a patient with HNF1A-MODY. Furthermore, miRNA has been reported as a gene regulator in diabetes [59].
CONCLUSION
The most important key element of diagnosing monogenic diabetes is identifying disease-causing genes that can classify patients into particular subgroups [60]. Different treatments can significantly affect the outcomes in these subgroups due to the efficacy of precision medicine in these subgroups with known etiology. Additionally, accurate diagnosis improves clinical prognosis and family genetic counseling. The phenotype of KCNJ11 neonatal diabetes depends on the severity of the mutation [61]. HNF4A-MODY patients with the p.R114W mutation have different phenotypes, showing decreased sensitivity to low-dose sulfonylurea, lower penetrance, and no effect on birthweight when compared to patients with other HNF4A-MODY mutations [62].
In Korea, the first genetic study for MODY was conducted in 2001 and focused on HNF1α [63]. Thereafter, various attempts were made to diagnose MODY patients [64,65]. Since 2015, whole exome sequencing has been utilized to identify candidate genes for MODY [66-68]. Recent studies using a targeted gene panel have been undertaken on Korean patients suspected of having monogenic diabetes [69]. This study reported that among 109 patients with probable monogenic diabetes, 23 (21.1%) carried pathogenically or possibly pathogenically mutations, with GCK-MODY being the most prevalent (50%). These results are similar to those of a comprehensive study on monogenic diabetes conducted in the United Kingdom [21]. According to a recent report from Japan, 39.2% were diagnosed with MODY; however, 18.4% showed de novo mutations contrary to the standard MODY criteria in this study [70]. Although standard MODY criteria include early onset (before age 25 to 35), absence of insulin dependence, absence of obesity and autoantibodies, and autosomal dominant inheritance for at least two generations [71], approximately 1% of MODY patients have glutamate decarboxylase antibodies, de novo mutations, and obesity, which make it challenging for the clinician to diagnose monogenic diabetes [72]. Approximately 60% to 80% of MODY’X’ was not identified in suspected monogenic diabetes patients, there is continuous attempt to find out unidentified genes for MODY. The precision diabetes treatment approach may still be challenging, and until genetic testing can be performed on every suspected patient with monogenic diabetes, the physician should prioritize patients with high suspicion through their biomarkers, probability score, or clinical characteristics. Regarding these difficulties, a proposed diagnostic algorithm for genetic testing in patients with diabetes could be helpful to the clinical community (Fig. 1).