Prader-Willi syndrome and growth hormone therapy: exploring the precise management of hypothalamic short stature: A review
Article information

Abstract
Prader-Willi syndrome (PWS) is a rare genetic disorder characterized by various clinical features linked to hypothalamic/pituitary gland abnormalities. Growth hormone deficiency is a prominent feature of PWS that results in poor linear growth and delayed development. This review discusses the evaluation and effects of growth hormone therapy (GHT) in PWS. Heterogeneity in growth hormone secretion patterns based on genotype and the potential for personalized GHT were explored. The benefits of GHT, including improvements in motor and cognitive development, growth, and body composition, are discussed in detail. Safety considerations for GHT initiation and response to GHT in adults with PWS are discussed, along with ongoing debates regarding the efficacy and safety. Although controversies persist, an evolving understanding of the long-term effects and safety of GHT underscores the need for comprehensive research in this field.
INTRODUCTION
Prader-Willi syndrome (PWS) is a rare genetic disorder caused by a lack of paternally expressed genes on chromosome 15q11-q13. Approximately 65% to 75% of PWS cases are caused by deletion of the paternal chromosome in that region, whereas maternal disomy 15 accounts for 20% to 30% of cases, and genomic imprinting defects cause 1% to 3% of cases. PWS presents a unique constellation of features, including hypotonia in infancy, hyperphagia, early onset morbid obesity, intellectual disabilities, hypogonadism, and behavioral abnormalities [1-4]. These symptoms appear to be associated with hypothalamic/pituitary gland abnormalities, and manifestations based on deficient pituitary hormones include the following [5-7]. (1) Growth hormone deficiency (GHD): insufficient production of growth hormone (GH) leads to poor linear growth and delayed physical development; (2) central hypothyroidism: reduced production of thyroid-stimulating hormone affects thyroid hormone release, leading to symptoms similar to those of primary hypothyroidism; (3) central adrenal insufficiency: adrenal hormone deficiencies can cause weakness, fatigue, low blood pressure, and a decreased stress response; (4) central hypogonadism: impaired production of gonadotropin-releasing hormone production leads to delayed sexual development and infertility; and (5) central diabetes insipidus: insufficient antidiuretic hormone production causes excessive thirst and frequent urination.
Early diagnosis and appropriate treatment are crucial for managing hormonal imbalances and improving the overall health and well-being of affected individuals. Among these symptoms, hypothalamic short stature with a lack of a pubertal growth spurt is particularly significant in PWS because of a deficiency of both GH and insulin-like growth factor 1 (IGF-1) [3,8,9].
In this review, short stature in PWS was explored, along with an exploration of ways to determine the effects, side effects, and duration of growth hormone therapy (GHT).
GROWTH HORMONE DEFICIENCY AND PRADER-WILLI SYNDROME
GHD is well recognized in PWS. Previously, GH stimulation tests were conducted to evaluate GHD in PWS. However, given the inherent nature of reduced GH secretion in the syndrome, once PWS is diagnosed, GHT is typically indicated after a certain age. Currently, in Korea, GHT for patients with PWS is covered by insurance, starting from the age of 2 years. Furthermore, most experts agree on initiating GHT before the onset of obesity, often setting the target age at < 2 years of age [10,11]. Although there may be slight variations across different studies, spontaneous impairment of GH secretion and low IGF-1 levels have been observed in nearly all patients with PWS. Approximately 80% of patients show a significant reduction in GHD levels, compared to the general pediatric population, through stimulation tests [11-15]. However, the frequency of GHD varies from 40% to 100% [12,15]. This discrepancy can be attributed to factors such as the type of stimulation test, non-standardized methods, country-specific diagnostic cut-offs, and the influence of stimulation test outcomes based on obesity [13,16-19]. In addition, it is important to consider the diverse normal ranges of GH levels according to age. Therefore, in PWS, comprehensive combined pituitary function and GH stimulation tests for hypothalamic-pituitary deficiency are essential. However, GHD may not align with the diagnostic cut-offs and could yield inconclusive results in PWS. Even in such cases, owing to the inherent nature of the disorder, GH supplementation should take precedence [11]. Indications and guidelines for GHT in PWS patients include comparisons between domestic, U.S. Food and Drug Administration, and GH Research Society recommendations (see Table 1 for details).
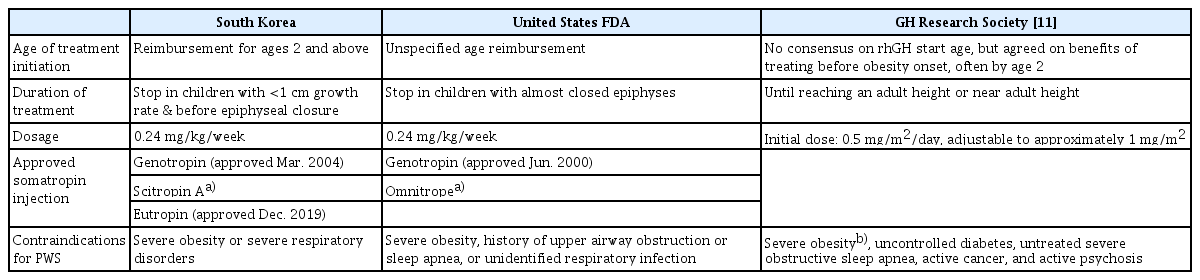
Indications for growth hormone injection therapy in Prader-Willi syndrome
THE ADVANTAGES OF GROWTH HORMONE THERAPY IN PRADER-WILLI SYNDROME
The advantages of GHT in PWS are as follows and have been demonstrated in numerous observational studies and RCTs [20-26]. (1) Enhanced motor development: a meta-analysis of five randomized trials involving 154 infants and children with PWS (aged 1.3 to 3 years) found that those receiving recombinant human growth hormone (rhGH) treatment demonstrated higher scores on standardized motor development tests (standard mean difference, 0.71; 95% confidence interval [CI], 0.38 to 1.03) [25]. One representative trial, focusing on infants and toddlers (29 participants aged 6 months to 3 years), revealed that rhGH therapy led to improved motor development during the initial year of treatment [27]. Standardized motor development scores on the Bayley Scales of Infant Development (BSID) increased by an average of 11.2% in the rhGH group, while the control group showed a decrease of 18.5%. (2) Enhanced cognitive development: in the same trial involving infants and toddlers, rhGH therapy also demonstrated improvements in cognitive development during the first year of treatment [26]. Standardized BSID scores for mental development increased by an average of 9.3% in the rhGH group, compared to a 2.9% decrease in the control group. However, in the earlier meta-analysis, which included six trials with 165 infants, children, and adolescents with PWS, cognitive scores were found to be similar between rhGH-treated children and untreated controls (standard mean difference, 0.2; 95% CI, –0.1 to 0.5) [25]. Notably, two of the trials in this analysis involved older children (average ages, 7 and 17 years, respectively), which may explain the lack of cognitive benefits in the overall analysis. (3) Improved growth and body composition: previous studies consistently show that rhGH therapy enhances linear growth and positively impacts body composition and obesity, regardless of whether treatment starts in infancy or later in childhood. A meta-analysis of nine randomized trials involving 328 patients demonstrated improvements in height and body mass index (BMI) in the rhGH group compared to the control group (mean difference in Z-score for height, 1.67 [95% CI, 1.54 to 1.81]; mean difference in Z-score for BMI, –0.67 [95% CI, –0.87 to –0.47]) [24]. Long-term data are needed to determine if rhGH therapy can increase adult height and potentially improve lipid profiles [28].
Observational studies and clinical trials have consistently reported similar benefits of rhGH therapy in infants and young children [22,23,29,30]. In a randomized trial involving 22 participants aged 5 to 32 months, the combination of rhGH and physical therapy increased muscle thickness, which correlated with improved muscle strength and motor performance compared to physical therapy alone in a control group [22,23]. Another trial (29 participants aged 4 to 37 months) showed that rhGH treatment resulted in improved mobility skill acquisition and body composition [30]. Starting rhGH treatment before 18 months of age has been linked to accelerated development of mobility skills, in line with expert recommendations to commence rhGH therapy at the time of PWS diagnosis, ideally within the first year of life [31].
The most substantial response to rhGH therapy in PWS children typically occurs within the initial 12 months of treatment [32]. However, sustained enhancements in height, bone density, and body composition have been observed with extended rhGH treatment, spanning up to 5 years [33]. Nevertheless, it is essential to emphasize that, despite extended treatment, full normalization of body composition may not be attainable. The chronological summary of various studies demonstrating the effects of rhGH treatment in pediatric Prader-Willi syndrome can be found in Table 2 [24,26,30,33-48].

Previous literature on the effects of growth hormone treatment in pediatric Prader-Willi syndrome
PRECAUTIONS FOR GROWTH HORMONE THERAPY IN PRADER-WILLI SYNDROME
Although GH replacement therapy has demonstrated general tolerability and has been extensively documented over the last 20 years, fatal events within the initial months of GH treatment have been reported in young patients with PWS. Such events are often associated with respiratory infections [49]. Despite the apparent improbability of a direct causal link between GHT and these fatalities, further investigations are warranted [11]. To ensure the safety of early initiation of GHT, thorough pretreatment assessments, including sleep studies, evaluations of the ear-nose-throat region, and thyroid function tests, are necessary. Regular polysomnography and continuous monitoring of the ear, nose, and throat area during therapy are recommended [50]. Adenoidectomy or tonsillectomy should be considered for issues related to lymphoid tissue. In addition, weight gain commences after the age of 2 years; therefore, strict monitoring for diabetes and blood glucose regulation is essential during GHT. Lifestyle adjustments are particularly necessary in cases of rapid weight gain [51]. GH treatment is not recommended for individuals with severe obesity, uncontrolled diabetes, or pre-existing severe scoliosis. Vigilant monitoring is essential in cases where treatment may exacerbate these risk conditions, alongside a comprehensive understanding of these potential risks [52,53].
RESPONSE TO GROWTH HORMONE THERAPY ACCORDING TO GENOTYPE
Differences exist in various clinical aspects between chromosome 15 deletion and maternal uniparental disomy (UPD) genotypes, which are genetic types representative of PWS [1,9]. These differences are also evident in the GH secretory pattern in response to GH stimulation. Regarding the GH secretory pattern, adults with PWS with del15q11-q13 exhibited higher growth hormone-releasing hormone (GHRH)+arginine-induced GH peaks than those with UPD15 [54,55]. Similar findings have been observed in pediatric age groups through various stimulation tests, such as clonidine, arginine, and insulin tolerance tests, which showed higher GH peaks in microdeletion cases than in UPD cases [13,56]. Furthermore, patients with the UPD15 genotype had a higher incidence of GHD than those with the del15q11-q13 genotype (80% vs. 25%). In line with these trends, both children and adults with the UPD genotype demonstrated better responses to GH stimulation than those with deletions [55].
However, it is unclear whether the growth-promoting and metabolic effects of GHT vary according to genotype. As there are differences in GH secretion and response to stimulation tests based on genotype in PWS, the response to and effects of GHT may also vary. However, the number of studies on this topic is limited. In a study comparing the effects of GH injection therapy in 65 Korean patients with PWS (21 UPD and 44 deletion types), no significant differences were found between the two genotype groups [57]. However, some studies have suggested that patients with specific genetic mutations may exhibit varying responses to GHT [8,13,54,56]. Therefore, further extensive research and investigation in this regard is imperative. In the future, GHT may evolve into a more personalized precision medicine tailored to individuals based on their genetic profiles.
GROWTH HORMONE TREATMENT FOR ADULTS WITH PRADER-WILLI SYNDROME
GH treatment for adults with PWS offers potential benefits, such as improved body composition, muscle strength, metabolic health, cognition, and quality of life [34,58]. However, safety concerns have been raised. Some studies have raised doubts regarding the efficacy and safety of long-term GHT in adult patients with PWS [11]. This debate centers on whether the gains in body composition from GHT are substantial enough to outweigh potential long-term uncertainties [59]. Critics argue that individuals with PWS are relatively protected from adverse metabolic effects and that intensive nutritional interventions can maintain healthy levels even without GH. GHT does not significantly reduce obesity-related issues [60-62]. Concerns have also arisen from elevated IGF-1 levels due to GHT, potentially raising cancer risk. Researchers have proposed that dietary and exercise interventions are more cost-effective for adult patients with PWS. Proponents of GHT assert that the natural persistence of hypothalamic dysfunction in PWS justifies continuous GH supplementation for consistent improvement in body composition, physical performance, and psychosocial function. They argued that an increase in IGF-1 levels is a biological effect without clear long-term consequences. The efficacy and safety of GHT in adult patients with PWS remain controversial, with research suggesting its benefits in various areas. Further studies are needed for a comprehensive understanding of the long-term effects and safety, necessitating patience to obtain conclusive results. The findings from the last 3 years are presented in Table 3 [63-65].
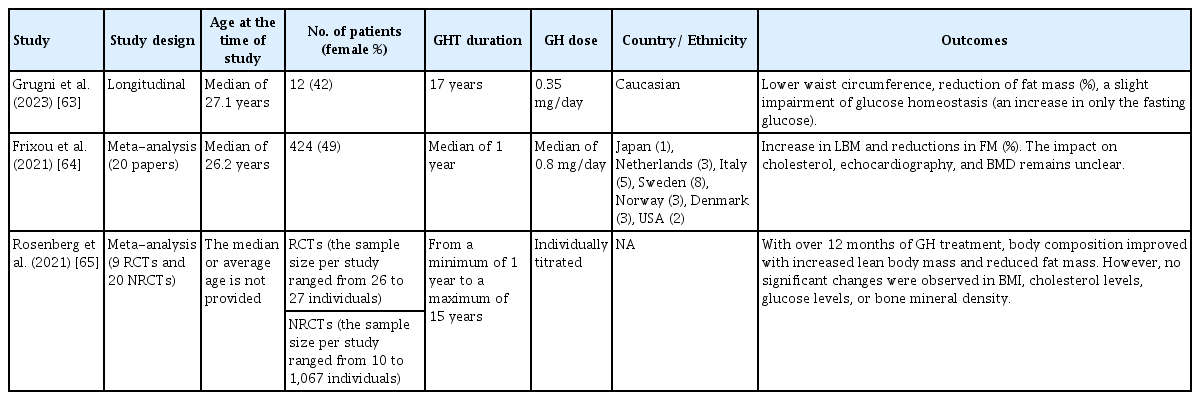
Recent studies on the effects of growth hormone treatment in adult Prader-Willi syndrome
INNOVATIVE APPROACHES AND RESEARCH DIRECTIONS
Ongoing research on PWS explores innovative treatment modalities beyond GHT. Interventions targeting appetite regulation, metabolic pathways, and behavioral aspects are being investigated to complement GHT and enhance the overall management of PWS. Collaborative efforts among clinicians, researchers, and geneticists are pivotal in driving these innovative approaches and expanding our understanding of the pathophysiology of PWS.
CONCLUSION
PWS is a complex disorder characterized by hypothalamic/pituitary abnormalities that lead to GHD, among other clinical manifestations. GH injection therapy has demonstrated beneficial effects on motor and cognitive development, growth, and body composition in individuals with PWS. However, the efficacy and safety of GHT in adults remains debatable. While some researchers have expressed doubts about its long-term benefits, others have emphasized the potential improvements in body composition and psychosocial function. A consensus on the benefits and risks of GHT in adults with PWS requires extensive long-term research. Future studies should address the divergence of opinions and provide a comprehensive understanding of the impact of GHT on this unique population. This evolving research landscape is expected to shed light on the optimal use of GHT and its potential contribution to the well-being of individuals with PWS.
Notes
No potential conflict of interest relevant to this article was reported.
AUTHOR CONTRIBUTIONS
Conception or design: AY.
Acquisition, analysis, or interpretation of data: AY.
Drafting the work or revising: AY.
Final approval of the manuscript: AY.