Molecular mechanisms of long bone growth and chondrocyte regulation: A narrative review
Article information

Abstract
Long bone growth is a fundamental determinant of final height. Growth, metabolism, and differentiation of chondrocytes, which are the key cellular players in this process, are regulated by systemic hormones, local factors, and cellular signaling pathways. This review provides an overview of the structural aspects of the growth plate, factors influencing chondrocyte function, and their impact on longitudinal bone growth. Systemic factors, including growth hormone, sex hormones, thyroid hormone, glucocorticoids, leptin, and insulin significantly affect chondrocyte proliferation and hypertrophy. Local factors, including transcription factors such as SRY-box 9 protein (SOX9), Runt-related transcription factor 2 (RUNX2), and bone morphogenetic proteins (BMPs), along with signaling pathways such as the Wnt pathway, play critical roles in chondrocyte proliferation and differentiation. These factors regulate gene expression, cell differentiation, and extracellular matrix synthesis. Additionally, Indian hedgehog (Ihh) and C-type natriuretic peptide (CNP) are involved in the complex signaling network governing chondrocyte function. Understanding molecular mechanisms underlying normal growth plate function has provided valuable insights into the genetic defects that impact growth and foundation for the development of effective therapeutic strategies for individuals with growth disorders.
INTRODUCTION
Long bone growth plays a pivotal role in determining overall height. The growth plate, a complex of cartilaginous structures situated between the epiphysis and the metaphysis of the long bones, orchestrates the process of endochondral ossification, which determines the linear growth of long bones [1-3]. Chondrocyte growth, metabolism, and differentiation are complex processes. The regulation of chondrocyte function is mediated by several systemic, local, and cellular signaling pathways. In this review, we summarized key regulatory factors and signaling pathways involved in chondrocyte functions, to understand how they affect chondrocyte growth and development.
STRUCTURE OF GROWTH PLATE
The growth plate can be stratified into three distinct zones based on the size, morphology, orientation, proliferative potential, and function of chondrocytes: resting, proliferative, and hypertrophic zone. The resting zone, a source of stemlike progenitor cells that restores the reservoir of proliferative chondrocytes, is located farthest from the primary ossification center and assumes responsibility for preserving the architectural integrity of the growth plate [4]. The proliferative zone, formed by cellular division originating from the resting zone, features flattened chondrocytes that undergo rapid replication, arrange longitudinally, and synthesize substantial amounts of extracellular matrix. Hypertrophic chondrocytes, a progeny of terminally differentiated chondrocytes from the proliferative zone, cease further proliferation, undergo hypertrophy in columns parallel to the axis of longitudinal elongation, and initiate the production of factors that trigger mineralization and vessel invasion. This, in turn, precipitates chondrocyte apoptosis, thereby, osteoblasts invade the hypertrophic zone and bone formation occurs [5]. At the end of puberty, the growth plate fuses with the epiphysis, and linear growth stops.
SYSTEMIC FACTORS AFFECTING THE GROWTH PLATE
Growth hormone (GH), sex hormone, thyroid hormone, as well as glucocorticoids affect chondrocyte proliferation and hypertrophy. Changes in any of these hormone axes can alter linear growth (Fig. 1).
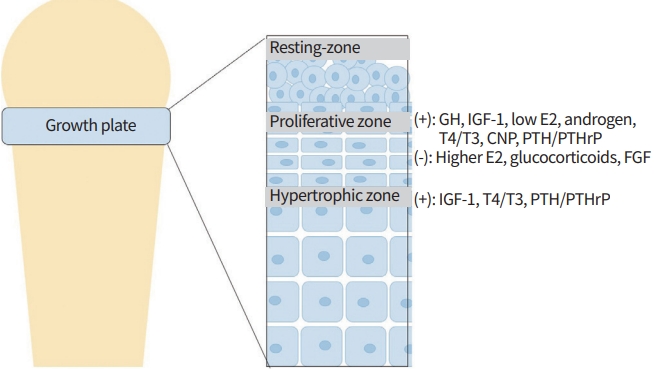
Structure of a long bone and the hormonal regulation of growth plate [13]. GH, growth hormone; IGF-1, insulin like growth factor-1; T4, thyroxine; T3, triiodothyronine; CNP, C-type natriuretic peptide; PTH/PTHrP, parathyroid hormone/parathyroid hormonerelated peptide; FGF, fibroblast growth factor.
GH–insulin-like growth factor (IGF) axis is a potent stimulator of linear growth. The original somatomedin hypothesis states that GH acts on linear growth by stimulating the production of IGF-1 in the liver [6,7]. The combined deficiency of liver-derived IGF-1 and the knockout of acid-labile substance reduce bioactive IGF-1 levels, thereby inhibiting linear growth and growth plate height [8]. In addition, administration of recombinant human IGF-1 demonstrated increased IGF-1 and significant improvements in linear bone growth in patients with inactivating mutations in the GH receptor [9]. According to the dual-effector hypothesis, GH directly recruits resting chondrocytes to proliferative chondrocytes and stimulates the local production of IGF-1, which acts in an autocrine/paracrine manner to increase chondrocyte proliferation and hypertrophy [10,11]. The IGF-1 signaling pathway is crucial for the formation of hypertrophic chondrocytes [12,13]. Growth plate-generated IGF-1 deficiency causes pre- and postnatal growth failure, characterized by disorganized columnar chondrocytes, decreased chondrocyte proliferation and hypertrophy, increased apoptosis, and delayed vascular invasion.
The pubertal growth spurt is induced by sex steroids, primarily estrogen. Estrogen appears to mediate this growth spurt by stimulating the estrogen-induced GH–IGF-1 axis, as well as by having direct effects on the growth plate [13-15]. Estrogen promotes skeletal maturation and epiphyseal fusion by exhausting the proliferative capacity of growth plate chondrocytes [16]. Additionally, estrogen stimulates the production of vascular endothelial growth factor, which promotes epiphyseal fusion. Androgens contribute to the growth spurt, partly through aromatization to estrogens in the growth plate [17]. However, even without aromatization, androgens stimulate linear growth through direct actions on proliferation and extracellular matrix synthesis in growth plate chondrocytes [18-20].
Thyroid hormones are essential for normal development and maturation of growth plate. Hyperthyroidism accelerates linear growth and endochondral ossification, while hypothyroidism delays these processes. Thyroid hormone has indirect effects on the growth plate through GH–IGF-1, and direct effects on chondrocyte proliferation, hypertrophy, vascular invasion, and columnar organization [21,22].
Excessive glucocorticoids can inhibit chondrocyte proliferation, hypertrophy, and extracellular matrix synthesis, leading to growth failure in children [23,24]. In addition to their direct effects, excess glucocorticoids may also involve other endocrine signals and have been associated with diminished GH secretion and alterations in bioavailable IGF-1 levels in some patients [25].
Leptin, which is primarily secreted by white adipose tissue and is elevated in obese children, enhances chondrocyte proliferation and differentiation [26]. Furthermore, leptin upregulates growth plate aromatase activity, leading to accelerated skeletal maturation [27,28]. In obese children, increased insulin concentration and activation of insulin receptors in the growth plate may cause accelerated linear growth and epiphyseal fusion, despite reduced GH production [13].
LOCAL FACTORS AFFECTING ON THE GROWTH PLATE
The growth plate formation and function are regulated by several transcription factors and multiple signaling cascades (Fig. 2). The SRY-box 9 protein (SOX9) is a key transcription factor for maintaining chondrocyte proliferation and to regulating differentiation [29,30]. SOX9 is abundant in chondrogenic drives the differentiation of mesenchymal stem cell into chondrocytes, pre-hypertrophic chondrocytes, and subsequent hypertrophic chondrocytes [31,32].

Runt-related transcription factor 2 (RUNX2) plays an important mediator of chondrocyte maturation and subsequent ossification [33]. While RUNX2 is expressed at low levels in proliferating chondrocytes, its expression increases in chondrocytes that are pre-hypertrophic, hypertrophic, and terminally differentiated [33,34]. Experimental studies have shown that RUNX2 regulates collagen X expression in hypertrophic chondrocytes and promotes the process of endochondral ossification [35].
Bone morphogenetic proteins (BMPs), which belong to TGF-β superfamily, are involved in most processes related to skeletal development and play critical roles in cartilage formation and maintenance [36]. BMP2, BMP4, and BMP7 contribute to chondrocyte differentiation by regulating SOX9 expression, and promote endochondral ossification by regulating RUNX2 transcription [37-39]. Furthermore, BMP3 promotes terminal hypertrophic chondrocyte maturation.
SIGNALING PATHWAYS AFFECTING ON THE GROWTH PLATE
Wnt signaling pathway plays a crucial role in regulating chondrocytes proliferation and differentiation. Wnt signaling can be classified as β-catenin-dependent and β-catenin-independent pathway (canonical and non-canonical pathway, respectively) [40]. In the canonical pathway, activation of Wnt signaling occurs when the secreted glycoprotein Wnt binds to the seven-transmembrane frizzled protein receptor and the lipoprotein receptor-related protein-5/6. Then, the cytoplasmic protein Disheveled is recruited along with the destruction complex consisting of Axin, glycogen synthesis kinase-3β, adenomatous polyposis coli, and casein kinase1, resulting in the suppressed β-catenin phosphorylation. Increased cytoplasmic β-catenin translocate to the nucleus where it forms a complex with T-cell factor/lymphoid enhancer factor and controls the transcription of the target genes SOX9 and RUNX2. The β-catenin signaling can regulate the mesenchymal stem cell differentiation, and the embryonic knock out of β-catenin gene leads to ectopic chondrogenesis [41]. The non-canonical signaling pathway includes the Wnt/Ca2+ signaling pathway, mitogen-activated protein kinase (MAPK), inositol triphosphate-intercellular calcium, the planar cell polarity (PCP) signaling pathway, and c-Jun N-terminal kinase (JNK), which activation occurs independently of β-catenin. Non-canonical signaling is also essential for cytoskeletal reorganization and chondrocyte differentiation. Loss of chondrocyte polarity and delayed endochondral ossification result from disruption of PCP protein localization [42].
Indian hedgehog (Ihh) is a secreted protein of the vertebrate hedgehog protein family produced by early pre-hypertrophic chondrocytes [36,43]. The binding of Ihh to transmembrane protein Patched (Ptc) blocks Ptc’s inhibition of Smoothened (Smo), a multichannel G protein-coupled membrane protein. This, in turn, activates the Gli gene family. Subsequently, Gli proteins enter the nucleus to control downstream gene expression, including SOX9, RUNX2, and parathyroid hormone-related protein (PTHrP) [36]. A negative feedback loop of Ihh and PTHrP maintains chondrocyte proliferation and differentiation [44,45]. Ihh induces PTHrP expression, which diffuses into the growth plate and enhances chondrocyte proliferation. Simultaneously, PTHrP acts on the parathyroid hormone (PTH)/PTHrP receptor to inhibit chondrocyte hypertrophy and suppresses Ihh expression [46,47]. Acrocapitofemoral dysplasia, caused by inactivating mutation of Ihh, is an autosomal recessive skeletal dysplasia characterized by postnatal onset of disproportionate short stature, small chest size, premature closure of epiphyses, and brachydactyly with small wide fingernails [48]. Homozygous or compound heterozygous inactivating mutations in PTH1R which encodes PTH/PTHrP result in Blomstrand dysplasia, a neonatal osteosclerotic dysplasia characterized by severe rhizo-meso-acromelic shortness of the limbs, polyhydramnios facial dysmorphism, and advanced skeletal maturation [49]; while heterozygous activating mutations in PTH1R cause Jansen type metaphyseal chondrodysplasia with delayed hypertrophic chondrocyte differentiation [50,51].
Transforming growth factor-beta (TGF-β) is involved in biological processes including development, cell growth, inflammation, and immune response [39,43]. Mutations in a member of the TGF-β superfamily have been reported to cause skeletal dysplasia [52]. The proteins of the TGF-β superfamily, including TGF-β, BMPs, growth and differentiation factors, and activin, bind to type II receptors and phosphorylate type I receptors. This activates either the Smad-dependent or Smad-independent MAPK signaling pathway [53]. In the Smad-dependent signaling pathway, complexes are formed by phosphorylated R-Smads and Smad4 form complexes, which move to the nucleus for regulating the expression of downstream genes including SOX9 and RUNX2 [54]. In the Smad-independent MAPK signaling pathway, phosphorylation of RUNX2 increases its transcriptional activity while activating JNK and p38 kinases, which alters the balance between BMP and TGF-β signaling and accelerates terminal chondrocyte differentiation [54,55]. In addition, TGF-βs downregulated Ihh while upregulate PTHrP, indicating the involvement of TGF-β signaling in the Ihh/PTHrP signaling [56].
Fibroblast growth factor (FGF) stimulates mitosis and angiogenesis. FGF activates the intracellular tyrosine kinase domain of the FGF receptor (FGFR), which subsequently activates downstream signaling pathways such as including RASMAPK and phosphoinositide 3-kinase (PI3K)-AKT, that are essential for cell proliferation, differentiation, and survival [36,43]. Several studies have demonstrated that FGF2, FGF9, and FGF18 enhance mesenchymal stem cell differentiation, chondrocyte proliferation, and extracellular matrix synthesis [57-59]. FGFR1/2 promotes in proliferating chondrocytes and promotes chondrocyte proliferation and differentiation in proliferating chondrocyte, whereas FGFR3 facilitates chondrocyte apoptosis in hypertrophic chondrocytes [60,61]. Activating mutations in FGFR3 inhibit chondrocyte proliferation and differentiation, which can cause achondroplasia, hypochondroplasia, thanatophoric dysplasia types 1 and 2 and severe achondroplasia with developmental delay, and acanthosis nigricans [60].
C-type natriuretic peptide (CNP) is a member of natriuretic peptide family. It widely expressed in hypertrophic chondrocytes on the growth plate [62,63]. The cellular effects of CNP, which are mediated by natriuretic peptide receptor 2 (NPR2), enhance the activation and synthesis of cyclic guanosine monophosphate (cGMP), resulting in the promotion of chondrogenesis through increased expression of cell adhesion molecules and glycosaminoglycan synthesis [64,65]. Acromesomelic dysplasia 1, also known as the Maroteaux type, is an autosomal skeletal dysplasia resulting from NPR2 loss-of-function mutations characterized by facial dysmorphism and severe short stature with normal intelligence [66]. Additionally, NPR2 heterozygous loss-of-function mutations account for 2% to 6% of idiopathic short stature, which is manifested as short stature with non-skeletal abnormalities [67].
Retinoic acid (RA), a derivative of vitamin A, is involved in various biological processes including limb pattering and development, and cell proliferation and differentiation [68]. The RA signaling pathway is activated through the binding of RA to a nuclear RA receptor (RAR)-retinoid X receptor (RXR) heterodimer complex, which is associated with RA response elements (RARE), leading to the regulation of transcription by recruitment of either nuclear receptor corepressors or nuclear receptor coactivators. Analyzing the effect of RA on growth plate regulation is challenging as it is involved in both stimulating and repression actions [69,70]. Furthermore, it seems that RA regulates other signaling pathways, such as Wnt, BMP, FGF, and Ihh [68].
The mammalian target of rapamycin (mTOR) pathway is a pivotal regulator of cell metabolism, proliferation, and survival and is involved in growth and homeostasis [71,72]. The mTOR pathway is activated by various factors, such as IGF, epidermal growth factors, glucose, amino acids, and oxygen, which activates PI3K-AKT signaling. In contrast, inhibited by adenosine monophosphate activated protein kinase inhibits the pathway when intracellular nutrients are depleted. The mTOR pathway is a crucial regulator of chondrocyte proliferation and hypertrophy. When mTOR in limb bud mesenchyme is disrupted, embryonic skeletal growth is impaired [73]. Deletion of mTORC1 resulted in a reduced extracellular matrix synthesis and delayed chondrocyte hypertrophy, while there was no effect on chondrocyte proliferation and apoptosis [73]. In contrast, deletion of mTORC2 resulted in delayed chondrocyte hypertrophy and shortened hypertrophic zone, although there were no defects in cartilage matrix synthesis, nor changes in the size of hypertrophic chondrocytes [74].
CONCLUSION
Endochondral ossification elongates long bones, which is a complex process affected by systemic hormones, local factors, and cell signals, thereby promoting the synthesis of extracellular matrix and expression of chondrocyte characteristics. Regulation of chondrocyte proliferation and differentiation depends on the interaction and coordination of several factors. Understanding the molecular mechanisms involved in normal growth plates can provide opportunities to discover genetic defects that interfere with growth and enable the development of efficient therapeutic strategies for patients with growth disorders.
Notes
No potential conflict of interest relevant to this article was reported.
AUTHOR CONTRIBUTIONS
Conception or design: EK.
Acquisition, analysis, or interpretation of data: EK.
Drafting the work or revising: EK.
Final approval of the manuscript: EK.
Acknowledgements
This research was supported by Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education (RS-2023-00243089).