Current advances in combining stem cell and gene therapy for neurodegenerative diseases
Article information
Abstract
Neuronal death is the common final pathologic pathway of various neurodegenerative diseases (NDs). Although central nervous system has little regenerative potential, it is expected that damaged neural tissue can be recovered by exogenous supplementation of stem cells; however, stem cell therapy cannot modulate specific causes of NDs, such as accumulation of extracellular amyloid peptides in Alzheimer’s disease. In contrast, gene therapy can deliver therapeutic genes to specific ND targets. Therefore, combining stem cell and gene therapy would have dual treatment mechanisms (regenerating damaged neural tissue and modifying specific causes of NDs) and lead to better clinical outcomes. In this review, we discuss various therapeutic genes that can be used to develop stem cell gene therapy for various NDs and the techniques for how therapeutic genes can be integrated into stem cells.
INTRODUCTION
Neurodegenerative diseases (NDs) are various incurable conditions in the central nervous system (CNS). Despite the different locations and symptoms of NDs, their common final pathological pathway is neuronal dysfunction and loss [1]. The human CNS has poor ability to repair damage because neurons cannot spontaneously regenerate from residual nervous tissues [2]. NDs result in permanent and progressive losses of various CNS functions such as cognition, memory, and/or motor functions. Accordingly, there are few effective regenerative treatments for NDs; the current focus is only on delaying their progress [3].
Stem cells have self-renewal and multi-lineage differentiation capacities [4]. Based on their abilities, they come into the spotlight as novel candidates for regenerative treatment of NDs. Various types of stem cells have been established from human organs including nervous system tissues and have shown significant treatment efficacy in animal models of NDs [5,6], and mesenchymal stem cells (MSCs) are the most advanced stem cell treatments for NDs [7]. However, recently neural stem cells (NSCs) have been identified from mammalian CNS and have been applied in preclinical and clinical ND trials [8]. NSCs are more promising than MSCs because of their spontaneous differentiation into neural cells [9]. However, stem cell therapies alone are still expected to have limited therapeutic effects in NDs because they cannot reverse specific ND processes. Therefore, disease-modifying abilities need to be added to the regenerative potential of stem cells to improve their treatment effects.
Gene therapy is a technique that uses therapeutic genes to treat or prevent diseases [10,11]. It is designed to introduce genetic materials into cells to modify genetic abnormality or to express beneficial proteins [12]. The combination of stem cell and gene therapy could be a technical breakthrough that increases the therapeutic efficacy of stem cells [13]. Therapeutic genes that can modify the specific deteriorated molecular pathways of NDs might give stem cell therapies regenerative and treatment functions simultaneously. Based on this concept, we focus in this review on candidate therapeutic genes that can be delivered to stem cell treatments and on methods of gene delivery.
THERAPEUTIC TARGETS AND CANDIDATE GENES FOR NDs
NDs can be divided into the acute and chronic according to the periods of damage. Stroke and spinal cord injury (SCI) are acute NDs that result from temporal damage to the CNS by vascular and physical accidents, respectively [14]; the duration of damage from acute NDs is from seconds to minutes. On the contrary, chronic NDs cause slow, progressive loss of particular or generalized neuronal subtypes, such as Alzheimer’s disease (AD), Huntington’s disease (HD), Parkinson’s disease (PD), and amyotrophic lateral sclerosis (ALS) [15]. Although the loss of neurons in the CNS is the common feature of acute and chronic NDs, there are different mechanisms of neuronal death. Because NDs have their own pathologies, the proper therapeutic genes for each disease should differ by disease type (Table 1) [16-71].
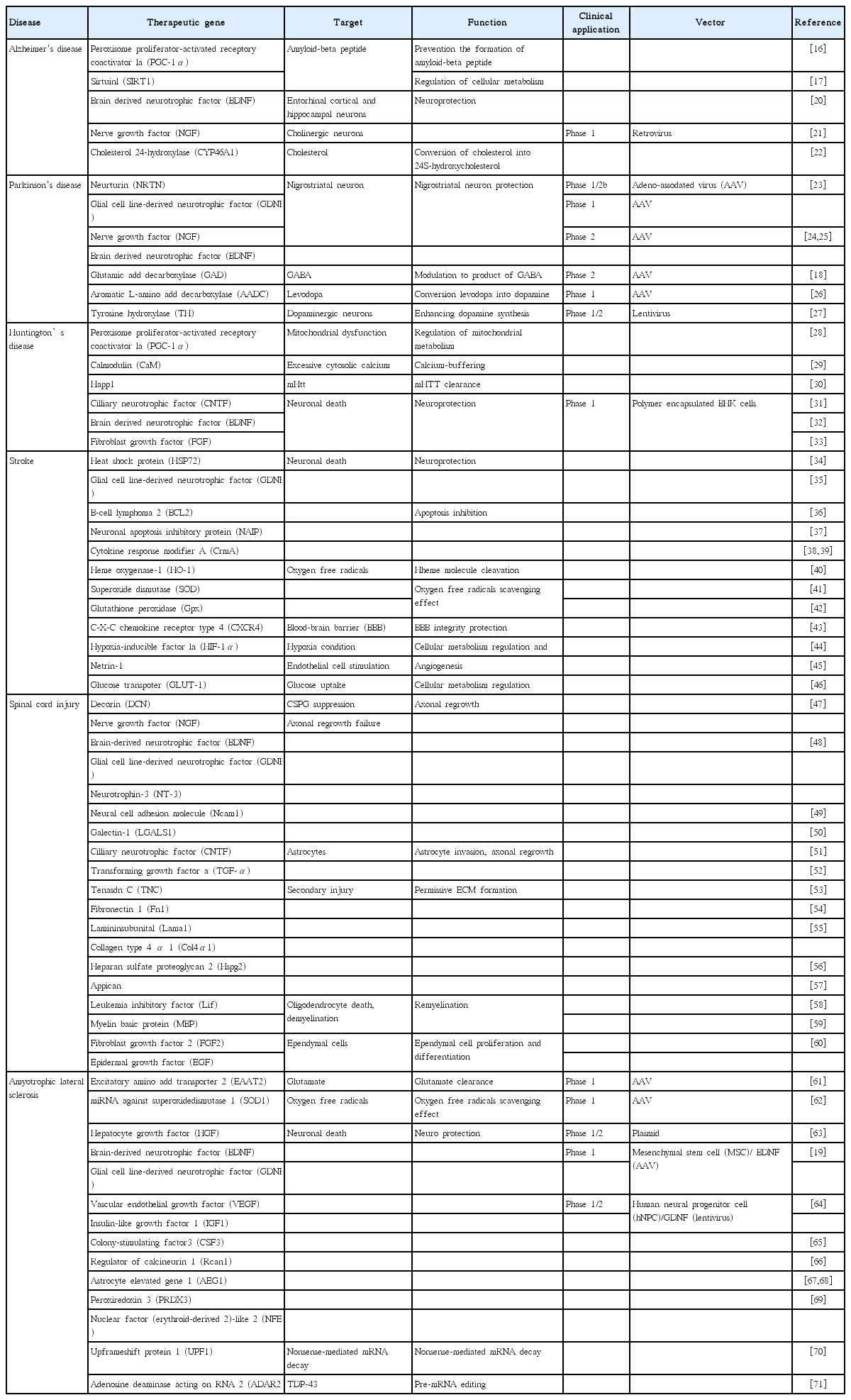
Therapeutic targets and candidate genes for NDs
Alzheimer’s disease
The pathologic features of AD include abnormal accumulation of extracellular amyloid (Aβ) peptides, formation of intraneuronal neurofibrillary tangles, extensive synaptic loss, and generalized cellular degeneration [72]. Among the various neuronal populations, the loss of the basal forebrain cholinergic neurons is particularly severe [73,74]; the loss of cholinergic neurons in AD correlates with functional severity of dementia, density of amyloid plaques in the brain, and amount of synaptic loss [75]. Therefore, gene therapies for AD have been developed to augment the function of degenerating cholinergic neurons or block neuronal death [76]. Nerve growth factor (NGF) or brain-derived neurotrophic factor (BDNF) gene delivery suppresses the death of the cholinergic neurons and elevates choline acetyltransferase function. Peroxisome proliferator-activated receptor γ coactivator 1α (PGC-1α) and sirtuin 1 (SIRT1) are expected to promote the nonamyloidogenic processing of amyloid precursor protein and preclude the generation of amyloidogenic Aβ peptides [16,17]. In contrast, cholesterol 24-hydroxylase prevents brain cholesterol accumulation, a risk factor for AD, by converting cholesterol into 24S-hydroxycholesterol.
Parkinson’s disease
Activities of mesencephalic dopaminergic neurons are specifically reduced in PD [18]. Because of its clear pathophysiology, gene therapy for PD is the most advanced in clinical trials (Table 1). To increase dopamine production, genes involved in dopamine neurotransmitter synthesis such as aromatic amino acid decarboxylase and tyrosine hydroxylase have been utilized [77-79]. Glutamic acid decarboxylase is expected to convert a subset of excitatory neurons to gamma-aminobutyric acid-producing inhibitory neurons whose activities are also reduced in PD. Adeno-associated virus (AAV) expressing NGF, glial cell-derived neurotrophic factor (GDNF), BDNF, and neurturin were designed to protect the degenerating nigrostriatum [80-82].
Huntington’s disease
HD is a hereditary triplet repeat disorder of the CNS in which certain gene sequences are mistakenly repeated [83]. The protein, mutant huntingtin (mHtt), is toxic, and it gradually damages neurons in the brain [84]. mHtt induces energy dysregulation by repressing transcription of PGC-1α, a regulator of mitochondrial metabolism. PGC-1α prevents loss of individual neuronal volumes and supports mitochondrial involvement. Therefore, mitochondria from HD patients show reduced calcium-buffering capacity and increased leakage of calcium into cytoplasm, which in turn causes excitotoxicity and aberrant calcium signaling. Calmodulin (CaM) is a regulatory protein that binds calcium and activates many enzymes upon calcium binding. Therefore, supplements of PGC-1α and CaM would protect mitochondria and buffer calcium effects, respectively. Happ1 is a recombinant antibody fragment that recognizes the polyP and P-rich domains of mHtt, and delivery of Happ1 could reduce mHtt aggregation. Genes of neurotrophic factors such as BDNF, ciliary neurotrophic factor (CNTF), and fibroblast growth factor (FGF) can enhance neuron survival.
Stroke
Stroke can be divided into ischemic and hemorrhagic. Blockage of blood vessels that supply oxygen and nutrients to the brain results in energy depletion and death of neural cells in the affected areas [85,86]. Gene therapy could be useful in treating stroke by stabilizing blood vessels (C-X-C chemokine receptor type 4 [CXCR4]), stimulating regrowth of blood vessels (hypoxia-inducible factor 1 [HIF-1] and netrin-1), or preventing neuronal death (HSP72, GDNF, B-cell lymphoma 2 [BCL2], neuronal apoptosis inhibitory protein [NAIP], and cytokine response modifier A [CrmA]) (Table 1). Although restoration of blood flow to an ischemic brain is essential to prevent irreversible brain injury, reperfusion may result in further damage by increased inflammation and oxidative stress. The production of toxic oxygen radicals by reperfusion might be reduced by the delivery of genes that enzymatically remove oxygen radical species (heme oxygenase-1 [HO-1], superoxide dismutase [SOD], and glutathione peroxidase [Gpx ]). Meanwhile, a glucose transporter gene (GLUT-1) would promote glucose uptake of neurons and prevent the energy depletion that induces apoptosis.
Spinal cord injury
SCI patients experience sudden loss of sensory, motor, and autonomic functions distal to the level of trauma [87,88]. The primary mechanism of SCI is necrosis of damaged neural tissues [89]; however, secondary mechanisms of SCI including ischemia, inflammation, and delayed apoptosis of neurons follow and worsen the functional losses [90,91]. Because the damaged lesions in SCI contain various exons that connect neurons, sensory organs, and muscles, reconnection could be a regenerative treatment for SCI. To improve axonal regrowth, many therapeutic genes have been tested, such as decorin (DCN), NGF, GDNF, neurotrophin-3 (NT-3), neural cell adhesion molecule (Ncam1), galectin-1, and CNTF (Table 1). In particular, DCN suppresses inhibitory chondroitin sulfate proteoglycan to promote axonal growth of neurons. Some therapeutic genes can target astrocytes or oligodendrocyte to make favorable environments for recovery. Transforming growth factor α (TGF-α) increases astrocyte invasion into damaged neural tissues and promotes axonal growth into lesions. Leukemia inhibitory factor (LIF) and myelin basic protein (MBP) are involved in the remyelination of oligodendrocytes that is important for stabilizing exons. Fibroblast growth factor 2 (FGF2) and epidermal growth factor (EGF) induce proliferation and differentiation of ependymal cells. Moreover, there are therapeutic genes that intend to reduce secondary injuries (i.e., fibronectin 1 [Fn1], laminin subunit α 1 [Lama1], collagen type 4 α 1 [Col4α1], heparan sulfate proteoglycan 2 [Hspg2], and appican). Using these therapeutic genes is expected to enhance axonal regrowth, neuroprotection, remyelination, and modulating microenvironments in SCI [92,93].
Amyotrophic lateral sclerosis
ALS is a ND that causes relentlessly progressive weakness of the arms, legs, and respiratory muscles [94,95]. Although there are no clear causes of ALS, 3% of patients have a familial form (FALS) that is phenotypically identical to the sporadic illness. FALS is caused by a mutation in SOD1 that makes excessive toxic oxygen radicals [19]; in addition, there are significant increases in the plasma levels of glutamate in ALS patients [96]. Based on the genotypes and phenotypes, gene therapy for ALS uses microRNA against SOD1 to induce degradation of SOD1 mRNA and a glutamate transporter gene (excitatory amino acid transporter 2 [EAAT2]) that clears excessive glutamates in microenvironments. The potential gene therapy for ALS may involve alteration in the mRNA processing (upframeshift protein 1 [UPF1] and adenosine deaminase acting on RNA 2 [ADAR2]) and inhibition of neuronal cell death (hepatocyte growth factor [HGF], BDNF , GDNF , vascular endothelial growth factor [VEGF], IFG1, colony-stimulating factor 3 [CSF3], regulator of calcineurin 1 [Rcan1], astrocyte elevated gene 1 [AEG-1], peroxiredoxin 3 [PRDX3], and nuclear factor [erythroid-derived 2]-like 2 [NFE2L2]). Especially, there are the clinical trials for ALS which apply stem cells expressing BDNF or GDNF [97].
GENE DELIVERY TECHNIQUES FOR STEM CELL GENE THERAPY
There are two major therapeutic gene-delivery methods in gene therapy. In vivo delivery entails directly injecting therapeutic genes into the body using viral or nonviral vehicles [98]. Ex vivo delivery loads cells with therapeutic genes, and then the cells that express the genes are injected into the body [99]. For stem cell gene therapy, therapeutic genes are transferred to stem cells ex vivo, and this results in transient or permanent expression of therapeutic genes within stem cells according to gene transfer methods (Fig. 1).

Gene delivery methods for stem cell gene therapy.
Gene transfer methods use viral or non-viral vectors that all have their own advantages and disadvantages (Tables 2, 3). In clinical trials, viral delivery methods have been utilized including adenovirus (AV), retrovirus (RV), lentivirus (LV), AAV, and herpes virus (HV) [100,101]. Viral delivery methods usually show high efficiency of therapeutic genes’ transduction and expression, although these can be affected by the genes’ sizes and vectors. AV is technically easy to produce and can carry relatively large genes, and it infects both dividing and nondividing cells. However, it has relatively high immunogenicity and the expression of therapeutic genes is essentially transient. However, RV has merits in the long-term stable expression of therapeutic genes if the genes integrate into the host genome; RV infects only dividing cells. The challenge is that integration of therapeutic genes into the host genome carries the risk of insertional mutagenesis. Due to the advantages of RV, it is the most used viral vector in clinical trials of gene therapy.
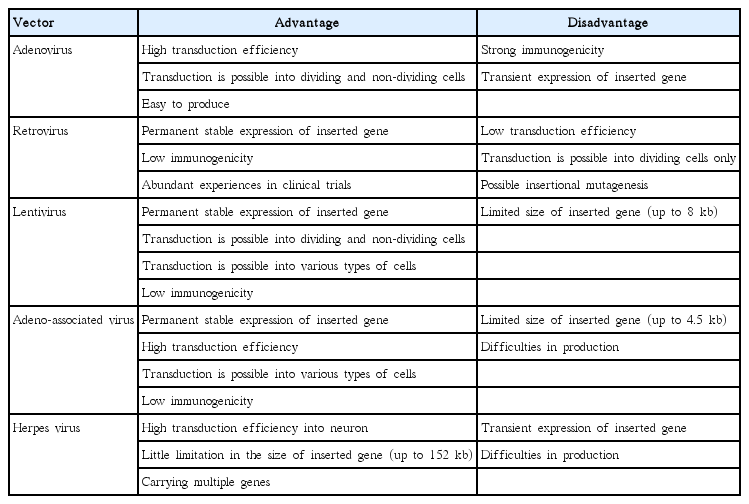
Advantages and disadvantages of viral therapeutic gene delivery methods
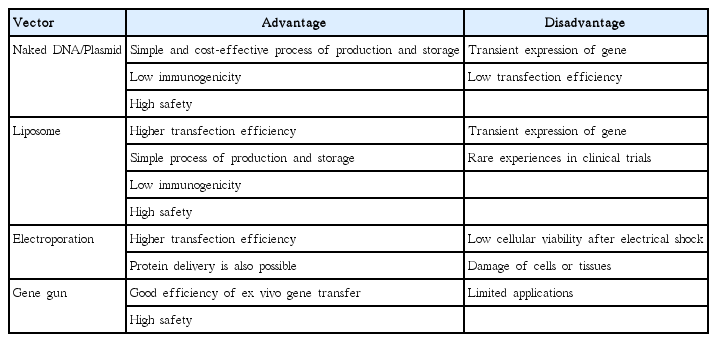
Advantages and disadvantages of non-viral therapeutic gene delivery methods
LV is a viral vector modified from human immunodeficiency virus (HIV) to have no disease-inducing activities and low immunogenicity. LV infects nondividing and dividing cells and delivers therapeutic genes to broader types of cells than RV. As with RV, LV shows long-term stable expression of therapeutic genes based on the genes’ integration into the host genome, and it can possibly result in insertional mutagenesis.
AAV infects both dividing and nondividing cells and has low immunogenicity. Long-time expression of inserted genes can be achieved with AAV. Although insertional mutagenesis is possible, site-specific integration of therapeutic genes into the host genome by AAV reduces the possibility dramatically. Limitation in the sizes of therapeutic genes and technical difficulties in the production are weak points of AAV.
HV can efficiently infect nondividing neurons in the CNS and carry multiple therapeutic genes at the same time. However, production of HV is relatively complex, and only transient expression of therapeutic genes is possible by HV. In many cases, viral delivery methods have safety concerns due to the permanent insertion of therapeutic genes into the host genome, which might provoke unwanted genetic mutations and tumorigenicity [102]. This risk is more important in stem cell gene therapy given that stem cells have been reported as possible sources of cancer development.
Nonviral delivery refers to plasmid-based delivery of therapeutic genes using chemical or physical stimulation [103]. The basic material of non-viral delivery is naked plasmid DNA (pDNA) containing therapeutic genes. The simplest application method is direct administration of naked pDNA without any chemical or physical assistances [104]. Various types of injection routes have been explored for naked pDNA including intravascular/intramuscular injection and inhalation [105]. However, the most disadvantage of it is low transfection efficiency. To improve the efficiency, mechanical or physical methods (i.e., electroporation and gene gun) have been applied to naked pDNA. Electroporation is brief electric pulses which induce the formation of transient pores in the membrane of target cells [106]. Such pores have little impact on the survival of cells but make functional ways through which pDNA can cross hydrophobic lipid bilayer [107]. The transfection efficiency of electroporation is reported as about 100- to 1,000-fold higher than that of direct administration of naked pDNA alone [108].
Gene gun is another physical approach to enhance the delivery of pDNA. Gene gun uses particle bombardment to shoot DNA-coated microscopic pellets through the cell membrane [107]. Recently a significant improvement in tissue penetration had been achieved using a newly designed gene gun, which allows longer gene expression of pDNA in subcutaneous tissues, such as muscle or tumor [109]. Liposome is a vesicular structure that is formed by the interaction and accumulation of lipids, in which pDNA is included [110]. Liposome is not a rigid formation but fluid entity that is versatile supramolecular assemblies. Based on its dynamic properties, it can fuse with the cell membrane to release pDNA into the cytoplasm. Moreover, liposome is relatively easy to manipulate, which make liposome used widely for the delivery of genes as well as drugs [111].
Compared with the viral methods, plasmid production is simpler, and there is little limitation of the size of the inserted gene. However, the efficiency of gene transfection and expression are lower than with the viral methods. Advantages and disadvantages of each nonviral method are summarized in Table 3.
CONCLUSION
Stem cell therapies have their own limitations in that stem cells by themselves cannot reverse the processes or correct the causes of NDs even though they regenerate damaged neural tissues. In contrast, gene therapy can correct genetic defects or express beneficial therapeutic genes that modify specific disease processes. Whereas many candidate therapeutic genes for NDs have been identified, their clinical uses as gene therapies are limited due to ineffective delivery and safety concerns. Combining stem cells and therapeutic genes might solve those issues in the gene therapy. Moreover, stem cells could regenerate neural tissue that cannot be repaired spontaneously. Although the methods of delivering therapeutic genes into stem cells is another technical issue, stem cell gene therapy might provide novel options for treating NDs that have more potent effects.
Notes
No potential conflict of interest relevant to this article was reported.
Acknowledgements
This research was supported by grants from Ministry of Food and Drug Safety in 2018 (18172MFDS182) and National Research Foundation (NRF-2016R1A5A2945889).