INTRODUCTION
Chronic myeloid leukemia (CML) is one of the disease entities included in myeloproliferative neoplasms (MPNs) by the ‘WHO classification of tumours of haematopoietic and lymphoid tissues’ [1]. TheBCR-ABL1 chimeric transcripts from the chromosomal translocation t(9;22) (q34.1;q11.2) is the molecular hallmark of CML and is the therapeutic target of tyrosine kinase inhibitors (TKIs) [2]. Depending on the breakpoints within the breakpoint cluster region gene (BCR) on 22q11 and the Abelson murine leukemia viral oncogene homolog 1 gene (ABL1) on 9q34, various forms of fusion transcripts have been identified [3]. In particular, three representative bcr have been described in the BCR gene: the major bcr (M-bcr; the area between exons 12–16), the minor bcr (m-bcr; between exons 1–2), and the micro bcr (μ-bcr; between exons 17–20). More than 95% of CML cases and approximately half the cases with acute lymphoblastic leukemia with t(9;22) haveBCR-ABL1 transcripts from the rearrangement at M-bcr, mostly the e14a2 (or b3a2) or e13a2 (or b2a2) types, producing a 210-kDa chimeric protein (P210). BCR-ABL1 transcripts from m-bcr produce a 190-kDa chimeric protein (P190), with the e1a2 type being the most common. The e1a2 type BCR-ABL1 is frequently detected at low levels in CML with e14a2 or e13a2 type BCR-ABL1 [4]. However, e1a2 BCR-ABL1 without other transcript types is rare in CML, accounting for approximately 1% of cases [5-9]. BCR-ABL1 transcripts (e19a2) from μ-bcr produce a 230-kDa chimeric protein (P230) and are also rare. Interestingly, recent studies have reported that the type of BCR-ABL1 transcripts affects the disease phenotype and outcome [10,11]. In particular, e1a2 BCR-ABL1 have been reported to be associated with prominent monocytosis, minimal basophilia, poor response to TKI, higher rates of disease progression to accelerated/ blast phases, and poor survival [5-9]. In this report, we describe a patient who had prominent monocytosis mimicking chronic myelomonocytic leukemia (CMML) but was diagnosed as having CML after the genetic workup that demonstrated t(9;22)/e1a2 BCR-ABL1 transcripts.
CASE REPORT
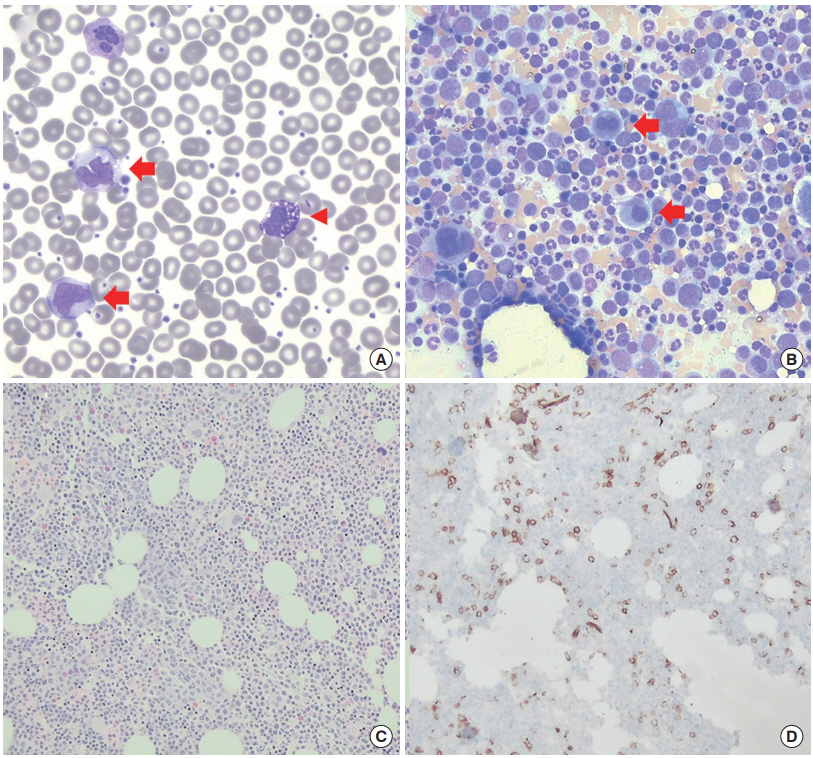
A 68-year-old woman was referred to the Division of Hematology-Oncology of Samsung Medical Center because of persistent thrombocytosis. She had been on follow-up for hypothyroidism. The complete blood counts (CBC) at referral showed leukocytosis and thrombocytosis with hemoglobin (Hb) 136 g/L, white blood cell (WBC) 19.470×109/L, and platelets 737×109/L (Table 1). The differential counts of WBC were band neutrophils 1%, neutrophils 56%, eosinophils 1%, basophils 4%, lymphocytes 15%, and monocytes 23% (absolute monocyte count [AMC], 4.478×109/L) (Fig. 1A). Neutrophils did not show definite dysplasia. The results from biochemical analysis were normal. Bone marrow (BM) study and related tests were performed with written informed consent from the patient and revealed hypercellular BM with 80% cellularity with increased granulopoiesis (without dysplasia) and megakaryopoiesis with dysplasia (megakaryocytes with separate nuclei and mononuclear forms) (Fig. 1B, C). Blasts were counted 4% on the aspirate smear. On the other hand, the CD34 immunostain on the biopsy section showed 5% to 10% CD34+ blasts (Fig. 1D). Conventional chromosome analysis using BM cells revealed 46, XX, t(9;22) (q34.1;q11.2)(20) (Fig. 2A), and fluorescence in situ hybridization (FISH) using Vysis LSI BCR/ABL Dual Color, Dual Fusion Translocation Probe (Abbott Molecular, Des Plaines, IL, USA) showed BCR and ABL1 gene rearrangement in 99% of interphase nuclei: nuc ish 9q34.1 (ABL1x3), 22q11.2 (BCRx3) (ABL1 con BCRx2)(198/200) (Fig. 2B). Multiplex reverse transcriptase polymerase chain reaction (PCR) using HemaVision-28N (DNA Diagnostic, Risskov, Denmark) showed minor (P190) BCR-ABL1 transcripts of the e1a2 type (Fig. 2C). Major (P210) BCR/ABL1 transcripts (including e14a2 or e13a2 types) or micro (P230) BCR-ABL1 transcripts (including e19a2) were not detected. Sanger sequencing of the BCR-ABL1 transcripts in the patient confirmed the junction with in-frame fusion of exon 1 of BCR and exon 2 of ABL1, without additional nucleotide changes (primer sequences available upon request) (Fig. 2D). Molecular genetic tests for driver gene mutations in MPN (JAK2 V617F, JAK2 exon 12, MPL exon 10, and CALR exon 9 mutations) were performed using peripheral blood leukocytes and were all negative. Collectively, she was diagnosed with CML, BCR-ABL1-positive, in chronic phase. A review of serial CBC before the BM study demonstrated that isolated monocytosis preceded the abnormalities in other parameters of CBC (Table 1). The follow-up CBC after 4 weeks of dasatinib medication (100 mg daily) showed Hb 106 g/L, WBC 2.590×109/L, and platelets 79×109/L.
DISCUSSION
According to the current World Health Organization classification of hematologic malignancies, there are three major disease categories in myeloid neoplasms other than acute myeloid leukemia: MPN, MPN/myelodysplastic syndrome (MDS), and MDS [1]. The MPN category includes CML (BCR-ABL1-positive), chronic neutrophilic leukemia (CNL), polycythemia vera (PV), and primary myelofibrosis (PMF). MPN/MDS includes CMML and atypical CML (BCR-ABL1-negative). The diagnostic criteria for each disease entity includes the presence of proliferative and/or dysplastic hematologic features (with predominantly affected lineages) and genetic aberrations. In particular, CMML is characterized by the presence of persistent (≥3 months) peripheral blood monocytosis (≥1.000 ×109/L and ≥10% of WBC) and dysplasia involving ≥1 myeloid lineages. While t(9;22)/BCR-ABL1 is required for the diagnosis of CML, the absence thereof is a prerequisite for the diagnosis ofthe other disease entities including CMML.
The patient described in this report had persistent monocytosis with AMC 4.478×109/L (23% of WBC) along with thrombocytosis in peripheral blood without significant leftshifted neutrophils (1%), suggesting the possibility of CMML (Table 1, Fig. 1A). However, the chromosome and FISH studies demonstrated t(9;22)(q24;q22) and BCR-ABL1 rearrangement, respectively, indicating CML. Multiplex PCR revealed e1a2 type BCR-ABL1 transcripts, which is very rare in CML. Of interest,the observation ofthis rare type oftranscripts in this patient was in line with the manifestation with prominent monocytosis [5-9]. On the other hand, while CML with e1a2 BCR-ABL1 has been shown to be accompanied by minimal basophilia, our patient had apparently increased basophils at diagnosis (7.788×109/L). It is also reported that CML with e1a2 BCR-ABL1 is associated with a high frequency of disease progression with increasing blasts. In our patient,the blast % on the aspirate smear was less than 5%, but CD34 staining on the biopsy section revealed >5% CD34+ cells (Fig. 1D). The patient is currently under treatment with dasatinib, a 2ndgeneration TKI. Close monitoring is warranted, due to the association of e1a2 BCR-ABL1 with poor response to TKI therapy and rapid disease progression.
While CML with e1a2 (P190) BCR-ABL1 is associated with prominent monocytosis and poor prognosis, CML with e19a2 (P230) BCR-ABL1 from the rearrangement involving μ-bcr is associated with prominent neutrophilia, mimicking CNL (thus, is also referred to as neutrophilic CML), and with indolent course [12,13]. Pane et al. [12] suggested that the transcription silencer elements located in the intronic sequences within M-bcr of BCR be the molecular basis of these different genotype-phenotype correlations [14]. Monocytosis also occurs in other MPN diseases. Barraco et al. [15] reported that 21% of patients with PV had monocytosis. Interestingly, they found significantly higher frequencies of tet methylcytosine dioxygenase 2 (TET2) and serine and arginine rich splicing factor 2 (SRSF2) gene mutations in PV with monocytosis than in PV without monocytosis. Since TET2 and SRSF2 are two of the most commonly mutated genes in CMML, this finding suggests that monocytosis in PV, and possibly CML, could be explained by CMML-associated mutations [16]. Monocytosis is also observed in PMF and was reported to be an independent predictor of inferior survival [17].
A review of the serial CBC of the patient showed that the mild increase of monocyte percentage in the WBC differential counts (8.6%) was the first flag to note (on November 30, 2015), followed by AMC (1.048×109/L, 15.1%) (on November 28, 2016) (Table 1). Until then, Hb, WBC, and platelet counts were within the reference ranges. Leukocytosis and thrombocytosis were detected on December 6, 2017 and progressed until the referral for BM study on January 18, 2019. These findings suggest that the increase of monocytes might be the first hematopathologic change in CML with e1a2 BCR-ABL1. Since the genetic workup forthe diagnosis of MPN can be performed using WBC in peripheral blood without invasive BM study, suspicion of the disease from CBC findings and timely genetic workup are importantfor early diagnosis and management.
In summary, we herein described a patient with CML from e1a2 (minor, P190) BCR-ABL1 transcripts, which is rare and associated with prominent monocytosis. Careful interpretation of CBC and timely genetic workup to determine the precise type ofBCR-ABL1 transcripts are critical forthe diagnosis and stratification of this rare subtype of CML with distinct genotype-phenotype correlations including poor prognosis.