Arginine methylation as a key post-translational modification in skeletal muscle homeostasis: a review
Article information

Abstract
Arginine methylation mediated by protein arginine methyltransferases (PRMTs) has emerged as a key post-translational modification of histone or nonhistone substrates. It involves or controls signaling pathways or gene expression implicated in diverse processes, including muscle regeneration and metabolic homeostasis. Reciprocally, loss of skeletal muscle mass and function related to aging or other pathological conditions could be related to the secondary chronic diseases, for example, metabolic syndromes, chronic inflammation, or cardiovascular diseases. Thus, understanding the pathways that regulate muscle homeostasis is critical to develop therapeutic strategies for preventing muscle loss and related secondary chronic diseases. Recent in vivo research using gene-targeting mouse models have advanced our knowledge about the role of several PRMTs in muscle regeneration and metabolic controls. In this review, we will focus on the recent discoveries on the in vivo function of PRMTs in muscle homeostasis.
INTRODUCTION
Skeletal muscle is the largest organ in humans, constituting about 40% of body weight in an adult, and plays critical roles in locomotion, energy expenditure, and glucose disposal [1-3]. Thus, alterations in metabolic characteristics as well as mass or strength of skeletal muscle affect the whole body’s energy metabolism and health [4]. Skeletal muscle from healthy adults displays a resilient adaptation in energy metabolism and contractile functions in response to demands like exercise, hormones, or nutritional states. Impairments in muscle metabolism and function have been implicated in metabolic pathologies, such as insulin resistance, glucose intolerance, and obesity [2,5,6].
Aging is accompanied by a progressive loss of muscle mass and function known as sarcopenia, which contributes to the reduced functional capacity and an increased risk for developing secondary chronic diseases, such as metabolic diseases, chronic inflammation, or cardiovascular diseases [2,7-9]. Thus, research efforts are focused on understanding the underlying mechanisms of muscle aging and to develop strategies to prevent muscle loss and weakness. One of the key factors contributing to muscle aging seems to be related to decreased muscle regenerative capacity accompanied by increased fibrosis. Thus, understanding the regulatory mechanisms of muscle stem cell function is critical for preventing muscle dysfunction.
Post-translational modifications such as phosphorylation, ubiquitination, and lysine methylation of diverse regulators have been shown to play critical roles in a variety of biological processes, including muscle regeneration and metabolic homeostasis [10]. Arginine methylation is a newly emerging post-translational modification of histone or nonhistone proteins. Protein arginine methyltransferases (PRMTs) catalyze mono-methylation and symmetric or asymmetric dimethylation of arginine residues on both histone and non-histone substrates, thereby modulating diverse signaling pathways and expression of genes involved in diverse biological processes, including muscle regeneration and muscle metabolism control [11-13]. PRMTs can be classified as type I, which catalyze asymmetric arginine dimethylation (PRMT1, PRMT2, PRMT3, PRMT4, PRMT6, and PRMT8), type II, which generate symmetric dimethyl-arginine residues (PRMT5 and PRMT9), or type III, which generate monomethyl-arginine residues (PRMT7) [11,12]. Several recent publications have advanced our understanding about the importance of several PRMTs in the control of muscle regeneration and function. This review focuses on the recent advances regarding PRMT function in controlling muscle stem cell function and controlling muscle metabolism.
THE ROLE OF ARGININE METHYLATION IN MUSCLE REGENERATION
Skeletal muscle has a resilient regenerative capacity via stem cells, also called satellite cells. Skeletal muscle regeneration is a multi-step process, including activation of quiescent satellite cells, expansion of activated progenitors, and terminal differentiation and fusion of myoblasts into preexisting myofibers [14,15]. Upon activation, paired box 7 (Pax7)-positive satellite cells express key regulatory factors belonging to the myogenic basic helix-loop-helix (bHLH) family (myogenic differentiation 1 [MyoD], myogenic factor 5 [Myf5], myogenin, and myogenic regulatory factor 4). These factors are critical to regulate myoblast proliferation and differentiation [16,17]. MyoD family bHLH factors function as master regulators to induce the expression of elemental muscle-specific genes and myogenic differentiation in non-muscle cells, like fibroblasts [18,19]. The activation of MyoD is regulated through multiple mechanisms including heterodimerization of MyoD with its partner E proteins (E12 and E47) that can bind to the consensus DNA sequence called the E-box (CANNTG, where N is any base), which is found in the regulatory regions of many muscle-specific genes [20-23]. Signaling pathways like p38 mitogen-activated protein kinase (p38MAPK) and AKT promote myogenesis through the activation of MyoD-mediated gene expression via phosphorylation of several transcriptional regulators including E proteins and the chromatin-modifying enzyme switch/sucrose non-fermentable (SWI/ SNF) subunit Brahma-associated factor 60c (BAF60c) [24-28]. Early studies using C2C12 myoblasts have shown that PRMT4/coactivator-associated arginine methyltransferase 1 (CARM1) and PRMT5 promote myoblast differentiation by regulating myocyte enhancer factor 2C (Mef2c) or MyoD, respectively [29,30]. In myogenesis, MyoD cooperates with histone-modifying enzymes and ATP-dependent chromatin remodeling proteins, such as the Brahma-related gene 1 (BRG1) and BAF subunit of SWI/SNF complexes, to promote the expression of downstream muscle genes [31]. Interestingly, PRMT5 is co-recruited to the myogenin promoter region with BRG1 and MyoD, leading to gene expression [30]. We recently reported that PRMT7 stimulates MyoD-mediated myoblast differentiation through methylation of p38MAPK at arginine residue 70 [32]. Studies with satellite cell-specific deletion mouse models for PRMT1, PRMT4, PRMT5, and PRMT7 have underlined the importance of arginine methylation in muscle stem cell function. PRMT4 regulates Myf5 induction through methylation of Pax7 during asymmetric division of satellite cells, which is critical for activation of satellite cells [33]. PRMT5 regulates muscle stem cell proliferation through silencing the cell cycle inhibitor p21 [34]. PRMT5 deficiency causes reduced H3R8me2s levels at the p21 promoter region, thereby enhancing its expression and leading to reduced proliferation of activated progenitors. PRMT1-deficient satellite cells exhibit enhanced proliferation with defective terminal differentiation through impaired regulation of MyoD [35]. PRMT1 regulates sineoculis homeobox homolog 1 (Six1), a coactivator of MyoD, through arginine methylation, which is critical for MyoD-mediated terminal differentiation. Lastly, it has been shown that PRMT7 controls the regenerative capacity of muscle stem cells in age-related manner. Adult mice null for PRMT7 or with conditional deletion of PRMT7 in satellite cells exhibited significantly impaired muscle regeneration due to premature cell cycle exit and cellular senescence [36]. PRMT7 ablation reduces the level of DNA methyltransferase 3b (DNMT3b), leading to the increased expression of p21 and cellular senescence in activated satellite cells. Based on these studies, it can be concluded that arginine methylation is critical for maintaining muscle stem cell function (Fig. 1). However, few PRMT substrates are known, and the upstream modulators of these enzymes or crosstalk between these enzymes are entirely unknown. Therefore, further studies are required to elucidate the defined mechanisms and function of PRMTs in maintaining muscle regeneration.
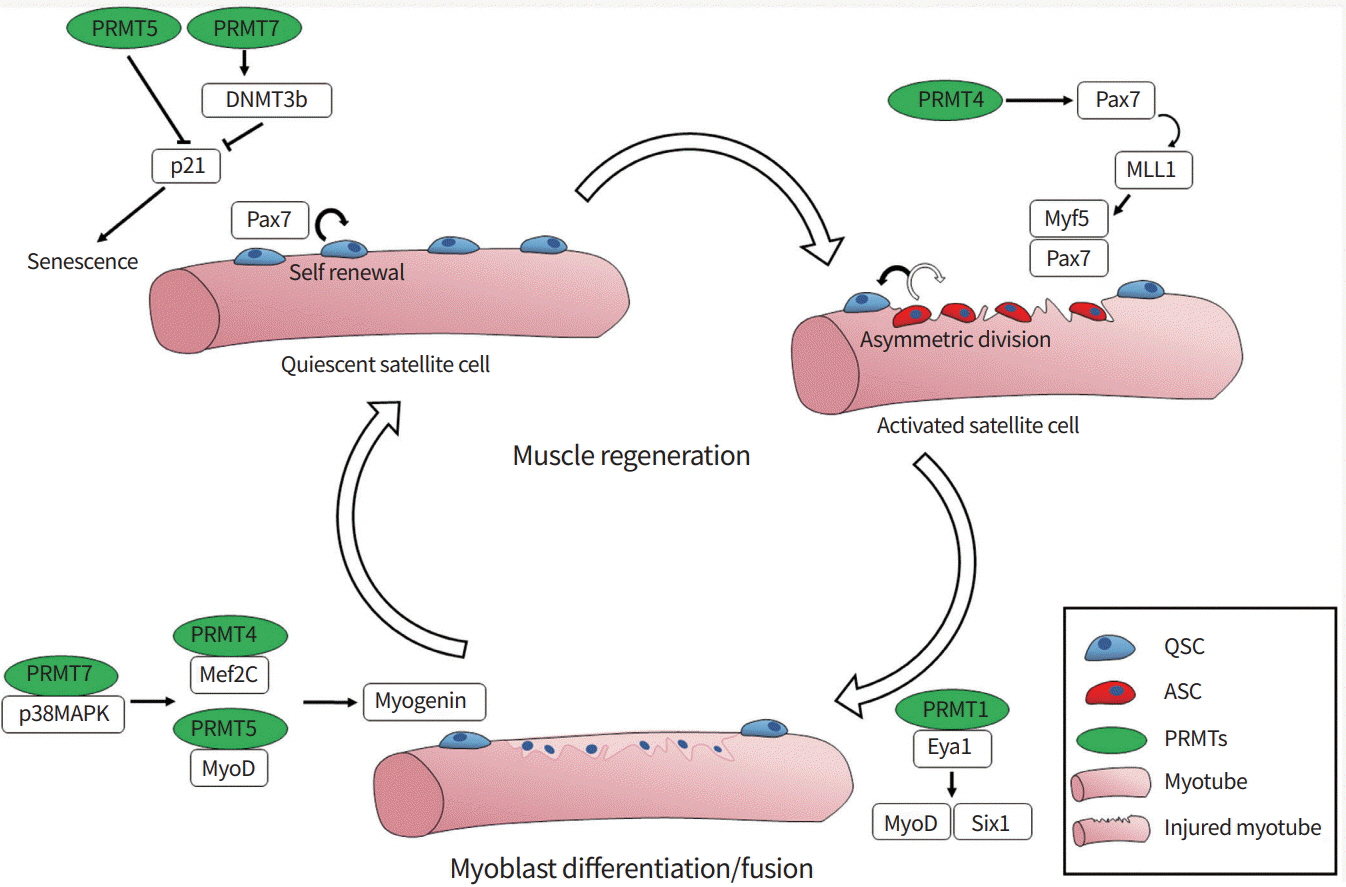
A scheme summarizing the role of protein arginine methyltransferases (PRMTs) in skeletal muscle regeneration. PRMTs play critical roles in muscle regeneration through modulating muscle-specific gene expression. PRMT5 and 7 play critical roles in preventing the senescence of satellite cells by suppressing the expression of the cell cycle inhibitor p21. Furthermore, PRMT1, 4, 5, and 7 regulate the activities of muscle-specific transcription factors such as paired box 7 (Pax7), myogenic differentiation 1 (MyoD), or myogenin, thereby promoting myogenesis. DNMT3b, DNA methyltransferase 3b; MLL, mixed lineage leukemia; Myf5, myogenic factor 5; p38MAPK, p38 mitogen-activated protein kinase; Mef2C, myocyte enhancer factor 2C; Eya1, EYA transcriptional coactivator and phosphatase 1; Six1, sineoculis homeobox homolog 1; QSC, quiescent satellite cell; ASC, activated satellite cell.
THE ROLE OF ARGININE METHYLATION IN SKELETAL MUSCLE METABOLISM
Recent studies have proposed critical roles of PRMTs in the control of skeletal muscle metabolism. Skeletal muscle exhibits a remarkable adaptation capacity in energy metabolism and contractile functions, and responds to various stimuli like exercise, hormones, or nutritional states. Skeletal muscle metabolism is modulated by glucose transport, mitochondrial biogenesis, and protein turnover [2,5,6,37]. Skeletal muscle mass is determined by the balanced regulation of protein synthesis and breakdown [38]. Chronic catabolic conditions, such as fasting or denervation, trigger elevated protein degradation and autophagy, thereby leading to muscle wasting. Excessive catabolic pathway signaling is triggered by enhanced AMP-activated protein kinase (AMPK) and p38MAPK activation, accompanied by induction of the proteolytic regulators Forkhead box O (FoxO), muscle-specific RING finger protein 1 (MuRF1), and atrogin [39-41]. The expression of PRMT1 and 4 is altered in denervation-induced atrophy or muscular dystrophic muscles, suggesting that these PRMTs might be involved in the atrophy process [42]. Consistently, recent studies have reported that these PRMTs are implicated in controlling the autophagy-proteosomal degradation that is critical for muscle mass control [43]. Muscle-specific ablation of PRMT1 causes muscle atrophy via PRMT6/FoxO3-mediated nuclear accumulation and enhanced autophagy [44]. PRMT4 also contributes to the autophagic process via FOXO3 methylation in atrophy-induced skeletal muscle [45]. Furthermore, PRMT4-dependent histone arginine methylation is also an essential nuclear event through AMPK-S-phase kinase associated protein 2 (SKP2)-CARM1 for autophagic induction after nutrient starvation [46]. The in vivo function of PRMTs in controlling anabolic pathways is currently unclear. Several in vitro studies on skeletal muscle cells have proposed potential roles of PRMT1 and PRMT4. PRMT1 regulates the insulin receptor (IR)/insulin receptor substrate-1 (IRS-1)/phosphatidylinositol 3-kinase (PI3-K) pathway involved in glucose transport in L6 skeletal muscle cells [47], while PRMT4 is necessary for the expression of genes involved in glycogen metabolism in skeletal muscle cells [48]. Further studies are required to determine the in vivo role of PRMTs in glucose metabolism and anabolic responses of skeletal muscle.
Skeletal muscle remodeling toward an oxidative metabolism is important for muscle function and whole-body metabolism. Recently, PRMT7 has been implicated in muscle oxidative metabolism through activation of p38MAPK/peroxisome proliferator-activated receptor gamma coactivator 1 α (PGC-1α) pathways, contributing to enhanced mitochondrial biogenesis and function [49]. PRMT7 deficiency causes reduced energy expenditure and induced age-dependent obesity. Previous studies have proposed a role of PRMT1 in inducing mitochondrial genes in non-muscle cells through methylation of p38MAPK and receptor-interacting protein 140 (RIP140) [50,51]. In skeletal muscle, the expression of PRMT1 and PRMT4 is induced by treadmill exercise [52], suggesting potential roles of these PRMTs in enhancing exercise-mediated oxidative metabolism. Further studies are required to define the role of other PRMTs that are expressed in skeletal muscle, the upstream inducers of PRMTs, or the epigenetic regulation of skeletal muscle homeostasis by PRMTs. Regardless of our limited current understanding, PRMTs have emerged as critical regulators in the control of skeletal muscle homeostasis (Fig. 2).
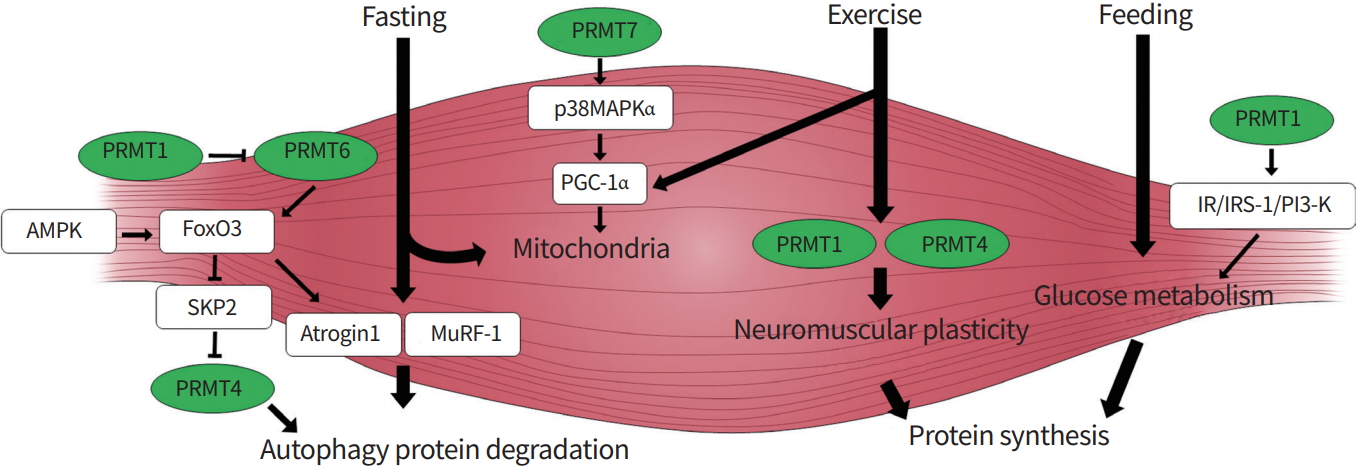
A model for the role of protein arginine methyltransferases (PRMTs) in the control of skeletal muscle metabolism. PRMTs (green) are implicated in the control of skeletal muscle homeostasis through different mechanisms involving phosphatidylinositol 3-kinase (PI3-K), Forkhead Box O3 (FoxO3), or p38 mitogen-activated protein kinase (p38MAPK) for skeletal muscle homeostasis. AMPK, AMP-activated protein kinase; SKP2, S-phase kinase associated protein 2; MuRF-1, muscle-specific RING finger protein 1; PGC-1α, peroxisome proliferator-activated receptor gamma coactivator 1α; IR, insulin receptor; IRS-1, insulin receptor substrate-1.
CONCLUSION
Muscle atrophy is caused by impaired regeneration capacities, metabolic processes, or protein turnover, leading to increased morbidity and mortality. In this review, we present the possibility of new diagnostic markers by understanding the cellular regulatory mechanisms in skeletal muscle maintenance. It is important to know the expression and activity of PRMTs as well as the changes in methylation of the target molecule and the downstream signaling systems in muscle repair and adaptation to stress. Understanding the roles of PRMTs in various steps during muscle cell regeneration and metabolism may provide new potential targets for countering muscle weakness and atrophy.
Notes
No potential conflict of interest relevant to this article was reported.
Acknowledgements
This work was supported by the grants from the National Research Foundation of Korea Grant funded by the Korean Government (NRF-2019R1I1A1A01062190 to Hyun-Ju Jeong).