INTRODUCTION
Metabolic syndrome (MS) is characterised by clusters of various metabolic abnormalities. It is one of the major risk factors for viral infections, and represents an emerging public health challenge. The latest clinical definition of MS according to the International Diabetes Federation (IFD), the American Heart Association (AHA), and the National Heart, Lung, and Blood Institute (NHLBI) is based on any of three of the five components, including abdominal obesity [1]. In clinical practice, the prevalence of MS is increasing steadily worldwide. It is estimated that approximately 20% to 25% of the adult population has MS worldwide. Moreover, in the United States, nearly 32% of adults were diagnosed with MS by 2000, increasing to 34% in the last few years [2]. Besides, MS is clearly a serious public health problem.
From a historical point of view, viral infection, specifically viral flu has not been kind to MS. Over the years, several cohort studies have reported a positive relationship between viral infections and MS or related metabolic disorders [3,4]. For instance, type 2 diabetes mellitus (T2DM), which is a characteristic disorder of MS, was one of the major risk factors for increased morbidity and mortality in patients infected with H1N1 Influenza, severe acute respiratory syndrome (SARS) coronavirus and Middle East respiratory syndrome coronavirus (MERS-CoV) [5]. According to recent reports emerging from the Centers for Disease Control and Prevention (CDC), patients diagnosed with MS and related disorders carry 10-fold higher risk of death following infection with coronavirus 2019 (COVID-19) [6]. It is well known that patients with MS and related metabolic disorders are at an increased risk of severe clinical outcomes and mortality not only in terms of virus infection but also several other infectious diseases [7].
Besides, MS and metabolic disorders such as obesity and T2DM are associated with dysregulated immune response against infections reducing the ability to detect antigens, blunting the healing process and delaying the recovery [8]. Notably, the host metabolic status has a direct impact on immune cells, such as natural killer (NK) cells and macrophages [9]. Animal studies also revealed that obesity dampens the immune response to infectious antigens, leading to severe outcomes post-infection. For instance, Honce and Schultz-Cherry [10] reported that the host innate and adaptive immunity are altered or dampened in obese individuals infected with influenza A virus (IAV) infection. It is disputed whether this weak immune response aggravates viral pathogenesis and viral spread and increases the risk of secondary infection. Several mechanisms have been suggested to explain the metabolic changes underlying the host susceptibility and the risk of viral infection. However, the precise mechanism and pathophysiology are still unclear. In this review, we will discuss this issue focusing on pro-inflammatory cytokines that might be the major factors exacerbating viral infection and resulting in severe clinical outcomes driven by specific intracellular events. We hope this review elucidates the factors underlying the increased morbidity and mortality in patients with comorbid MS and virus infection. Again, because the dysregulation of immune cells in pathogenesis of MS and other metabolic diseases has been extensively studied, our emphasis is on the effect of metabolic dysregulation of cytokine release and low grade-inflammation in viral infection.
PRO-INFLAMMATORY CYTOKINES
Since early years, it has been reported that MS is associated with increased levels of pro-inflammatory cytokines such as interleukin-6 (IL-6), tumour necrosis factor-α (TNF-α), and C-reactive protein (CRP) both in serum and adipose tissue [11]. It is mostly explained by the high levels of leptin and low levels of adiponectin as well as immune dysregulation [12]. The cytokine dysregulation in turn leads to systemic and chronic low-grade inflammation in MS and other related disorders such as obesity and T2DM [13,14]. Animal studies also confirmed that obesity increased the expression of IL-6, TNFα, and CPR early after infection of IAV, whereas increased inflammation was observed later in post-infection [11]. Similarly, human studies also revealed that patients with MS and obesity present with chronic low-grade inflammation due to increased pro-inflammatory cytokines associated with increased leptin and reduced adiponectin, which in turn leads to systemic effect [12,15,16]. These findings suggest that MS and other metabolic abnormalities are highly associated with metabolic inflammation and increased levels of pro-inflammatory cytokines.
However, virus infection per se is also the primary inducer of high level of cytokines via pro-inflammatory response even in normal host [17]. Furthermore, the impact of host metabolic inflammation and increased levels of circulating cytokines on viral infection, progress and spread as well as the role of pathophysiology in mortality and morbidity of patients is critical for improved disease management. Recent reports suggest that the cytokine storm is the common cause of death in COVID-19 and emphasize that patients with metabolic disorders including obesity and T2DM appear to be at a higher risk of severe clinical complications and even multiple organ failure (MOF) [18]. In addition, the high levels of serum IL-6, CRP, and creatinine accompanied by hyperglycosemia in patients with T2DM compared with non-diabetic patients are associated with more severe clinical complications and increased mortality among patients with COVID-19 as reported by Zhu et al. [19] from Wuhan University.
Overall, patients living with MS already present an increased level of circulating pro-inflammatory cytokines accompanied by systemic low-grade chronic inflammation. These pre-existing dysregulated conditions are conducive to viral infection, replication, spread, and progress in various ways. While pre-increased pro-inflammatory cytokines overlap with virus-induced cytokine release leading to more rapid cytokine storm, the impaired immune response facilitates viral infection. The excessive cytokine release induces cellular dysfunction leading to MOF [20-22].
PATTERN RECOGNITION RECEPTORS AS A PRELUDE TO INTRACELLULAR EVENTS
The inflammatory process, observed in MS, establishes itself systemically and is characterised by a chronic low-intensity reaction. The signalling pathways related to pattern recognition receptors (PRRs) are one of the main triggers of metabolic inflammation [23,24]. There are several types of PRR subfamilies, such as Toll-like receptors (TLRs), retinoic acid-inducible gene I-like receptors, C-type lectin receptors, nucleotide-binding oligomerization domain (NOD)-like receptors (NLRs), and DNA sensors [25]. The TLRs and NLRs are the two most characterised innate receptors underlying the progression of metabolic disease, which induces a downstream intracellular signalling cascade to release mediators of inflammation such as cytokines, chemokines, and co-stimulatory molecules [26,27]. Multiple PRRs have been implicated in the recognition of metabolic stress and initiation of inflammatory responses in various tissues [28]. The main ligand of PRRs in MS are metabolic stress and excessive nutrients. MS and related metabolic disorders trigger diverse events, such as endoplasmic reticulum (ER) stress, hypoxia, and lipotoxicity, which in turn promote the activation of pro-inflammatory pathways within the metabolic tissues, including adipose tissue, liver, pancreas, gastrointestinal tract, muscle, hypothalamus, and blood vessels through PRRs [29]. Thus multiple factors play a crucial role in MS-associated inflammation.
However, the “antigen-immune response-metabolic homeostasis” is highly dependent on each of the individual components. Since we have discussed the viral infections in patients with MS, it is appropriate that we also consider the various receptors associated with the infection. PRRs recognize the innate immune system, which is the first layer of host defence against infection. Upon infection, PRRs recognize viral proteins and RNA to trigger the appropriate protective immune response in normal hosts. However, in case of MS, PRRs have been persistently activated by metabolic stress, and are dysregulated eventually. Viral stimulation of altered PRRs triggers excessive inflammatory response via various signalling pathways ending in excessive cytokine release [30,31]. Several studies are investigating the role of additional PRRs both in MS and virus infection. However, further investigations are warranted given their key role in metabolic homeostasis and immune response to infection.
NF-κB RELATIONSHIP WITH METABOLIC SYNDROME AND VIRAL INFECTION
MS is associated with excessive energy sources and metabolic levels that in turn overload the capacity of metabolic tissues under metabolic stress. The metabolic stress overwhelms the nutrient levels leading to activation of intracellular events such as signalling pathways that in turn promote further metabolic abnormalities such as insulin resistance, cytokine production and immune cell recruitment. Nuclear factor-κB (NF-κB) is a well known inflammatory signalling pathway that is crucially involved in metabolic inflammation [32]. Further, the NF-κB signalling pathway mediates the immediate-early pathogen response, promoting pro-inflammatory cytokine release and inflammation, and regulation of cell proliferation and survival [33,34].
As mentioned previously, hyperlipidemia and hyperglycaemia in MS stimulate TLRs [35,36]. Saturated fatty acids are also believed to bind to TLR4, thereby activating IKK2 in metabolic tissues such as adipocytes, macrophages, and muscles leading to a reduction in insulin sensitivity [37,38]. Besides, overnutrition triggers ER stress. MS is also associated with dampened insulin signalling, which leads to persistently high levels of glucose. Under these conditions, pancreatic islets secrete higher levels of insulin, which requires appropriate folding of ER. Eventually, ER stress contribute to major beta-cell dysfunction due to the activation of inflammatory pathways such as NF-κB signalling pathway [39]. Such ER stress also occurs in other metabolic tissues such as liver and adipose, eventually triggering systemic inflammation [40,41]. Further, the increased nutrient levels committed to ER stress in hypothalamus results in activation of NF-κB signalling, which leads to central insulin resistance [35,42]. Furthermore, the nutrient overload and activation of NF-κB result in systemic inflammation and expression of various pro-inflammatory cytokines in MS and other metabolic disorders.
Second, the activation of NF-κB is critical to the antiviral immune response. NF-κB signalling occurs within minutes of stimulation and no protein synthesis is required. Moreover, NF-κB affects several critical steps in the host cell lifecycle, suggesting that the NF-κB pathway is an attractive target to the invading virus. Most often, viruses act via diverse mechanisms to evade and exploit the NF-κB pathway for viral replication [43]. The IAV uses NF-κB pathway for more efficient replication. Evidence suggests that the p65 NF-κB molecule activates influenza virus RNA transcription from the cRNA promoter [44]. Viral oncogenic proteins activate NF-κB in viral infection, which is extensively reviewed [45-47]. Upon infection with virus, the PRRs on immune cells recognize pathogen-associated molecular pattern, followed by interaction between TLR3 and Toll-interleukin-1 receptor domain-containing adapter inducing interferon-β (TRIF), and downregulation of adaptor myeloid differentiation primary response gene 88 (MyD88) by other TLRs [48]. NF-κB activation results in up-regulation of hundreds of genes including the expression of inflammatory cytokines that constitute a robust antiviral immune response [49].
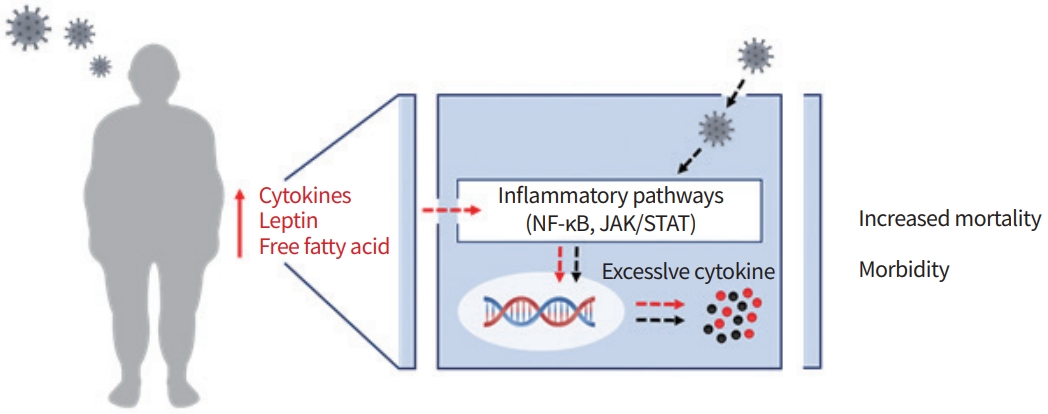
Obviously, appropriate immune response against viral infection during normal host NF-κB activation is mediated via adequate levels of inflammatory cytokines. However, patients with MS and other metabolic disorders have systemic low-grade inflammation, which dampens the immune response to persistent stimuli. Besides, they also carry increased levels of circulating pro-inflammatory cytokines. This overwhelming cytokine release results in severe clinical outcomes. Since NF-κB is a critical regulator of inflammation both in MS and viral infection, NF-κB is the potent target to control the cytokine release (Fig. 1). However, the tissue-specific roles and other aspects need to be investigated.
JAK/STAT SIGNALLING PATHWAY RELATIONSHIP WITH METABOLIC SYNDROME AND VIRAL INFECTION
Patients with MS present with a high level of serum leptin associated with systemic leptin resistance [50,51]. Upon binding with leptin and leptin receptor, the Janus kinase 2 (JAK2) is activated via auto-phosphorylation, involving the cytoplasmic tail of receptor [52], which in turn leads to phosphorylation of signal transducers and activators of transcription (STAT) 3 and STAT5 resulting in transcription of inflammatory cytokines (Fig. 1). Emerging evidence implicates dysregulation of JAK/STAT pathway signalling in obesity and metabolic disease. Impaired lipolysis, weight gain and adiposity in response to growth factor and leptin were observed in adipose-specific deletions in JAK2 knockout (KO) mice. Moreover, some studies also reported that the deletion of STAT2 or STAT5 in mice leads to insulin resistance [53]. By contrast, other studies reported increased insulin sensitivity under STAT4 deficiency in adipocytes [54]. This attenuation may be explained by the insulin resistance due to free fatty acids, suggesting that the loss of JAK2 leads the enhanced insulin sensitivity. These findings suggest that the JAK/STAT pathway is required for normal metabolic homeostasis.
Also, the IL-6/JAK/STAT3 signalling pathway is the critical subtype of the JAK/STAT pathway including IL-6. Both MS and viral infections are strongly associated with elevated levels of circulating IL-6. In MS, the adipose tissue including resident immune cells and adipocytes are the primary producers of IL-6, while infected and immune cells are the main producers in viral infection [55]. Recent studies suggest that the elevated levels of circulating IL-6 are the main indicator of cytokine storm and poor prognosis in COVID-19 [18,19]. The binding of IL-6 to its specific receptor (IL-6 receptor-subunit alpha IL-6R) induces the expression of IL-6/JAK/STAT3. Thus, IL-6 triggers the expression of inflammation-related downstream target genes [56]. The specific role of JAK/STAT pathway in metabolism is extensively reviewed by tissue-specific in vivo experiment [57,58]. Furthermore, the activation of JAK/STAT pathway by increased levels of leptin and IL-6 may be another major factor underlying the excessive cytokine release, thereby worsening the prognosis of viral infection in patients with pre-existing MS.
It is also suggested that JAK/STAT pathway is dysregulated in MS and related metabolic disorders [59]. Since JAK/STAT pathway is crucial for the regulation of inflammation, dysregulation of JAK/STAT pathway results in excessive cytokine release. IL-6 and leptin-related activation of JAK/STAT signalling may exacerbate the excessive cytokine release and systemic inflammation in patients infected with the virus in the context of metabolic dysregulation. Further evidence suggests that JAK/STAT inhibitors attenuate the viral infection by attenuating the excessive cytokine release in COVID-19. Clinical trials have been initiated with IL-6 antagonists in patients with COVID-19 [60]. Among JAK inhibitors, tofacinib is approved for use in autoimmune disease in which cytokines play a main role [61]. An interesting case was reported involving a woman with a 13-year history of ulcerative colitis who contacted SARS-CoV-2 and was treated with tofacitinib. After 2 weeks, her symptoms were improved even without hospitalization [62]. Besides, ruxolitinib, a JAK1/2 inhibitor, showed rapid clinical attenuation in patients with COVID-19 [63]. Similar studies using the JAK inhibitor, baricitinib, also showed clinical attenuation in COVID-19 patients [64-66]. Further studies investigating the benefits of JAK inhibitors on excessive cytokine release are still under way. Overall, the major risk factors carried by patients with MS increase the susceptibility to viral infection via excessive cytokine release and systemic inflammation, and JAK/STAT pathway represents a potential therapeutic target for controlling cytokine release.
CONCLUSION
In this review, we have briefly discussed the pathogenic factors that increase the risk of viral infection in patients diagnosed with MS. We focused our attention on excessive levels of proinflammatory cytokines that are regulated by NF-κB, JAK/STAT signalling pathways mediating metabolic inflammation. During MS, the dysfunctional adipose tissue-derived cytokines trigger inflammatory signalling pathways, which in turn result in the synthesis of additional inflammatory cytokines. Thus, patients living with MS and metabolic disorders such as obesity and T2DM carry increased levels of circulating cytokines and systemic low-grade inflammation. Upon viral infection, such patients with all pre-existing backgrounds, specifically dysregulated inflammatory signalling, leads to rapid viral replication and results in worse clinical outcomes (Fig. 1). Therefore, viral infection in patients with pre-existing MS and metabolic disorders require special care and management. Until now, no specific treatment strategy is available or suggested. Better understanding of disease pathogenesis and precise mechanism is necessary for accurate diagnosis and treatment. Here we suggest that the increased pro-inflammatory cytokine release is a risk factor, and therefore, targeting inflammatory signalling pathways represents a potential therapeutic target to control the cytokine release, and thereby prevent serious outcomes, such as increased mortality and morbidity.