Autoantibodies in central nervous system and neuromuscular autoimmune disorders: A narrative review
Article information

Abstract
The discovery of novel autoantibodies in neurological disorders contributes to a better understanding of its pathogenesis, improves the accuracy of diagnosis, and leads to new treatment strategies. Advances in techniques for the screening and detection of autoantibodies have enabled the discovery of new antibodies in the central nervous system (CNS) and neuromuscular diseases. Cell-based assays using live or fixed cells overexpressing target antigens are widely used for autoantibody-based diagnosis in clinical practice. Common pathogenic autoantibodies are unknown in most patients with multiple sclerosis (MS) and chronic inflammatory demyelinating polyradiculoneuropathy (CIDP). Novel pathogenic autoantibodies to aquaporin-4 and myelin oligodendrocyte glycoprotein (MOG) have been identified in neuromyelitis optica spectrum disorder and MOG antibody-associated disease, respectively. These diseases have clinical similarities to MS, but with the discovery of pathogenic autoantibodies, they are now recognized as distinct disease entities. Antibodies to paranodal membrane proteins such as neurofascin-155, contactin‑1, contactin‑associated protein‑1 in CIDP and muscle-specific kinase and low-density lipoprotein receptor–related protein 4 in myasthenia gravis were added to the profiles of autoantibodies in neurological disorders. Despite the relatively low frequency of seropositivity, autoantibody detection is currently essential for the clinical diagnosis of CNS and neuromuscular autoimmune disorders, and differential approaches to seropositive patients will contribute to more personalized medicine. We reviewed recent discoveries of autoantibodies and their clinical implications in CNS and neuromuscular disorders.
INTRODUCTION
The immune response protects the body from foreign substances. Excessive or insufficient immune responses can lead to immune disorders. Immune-mediated diseases of the central nervous system (CNS) or neuromuscular system cause antibody- or cell-mediated inflammation and tissue injury, which are potentially reversible through immunosuppression or modulation. Extensive research to define autoimmune pathogenesis and biomarkers has contributed to a better understanding of the diseases and resulted in a rapid expansion of the spectrum of autoimmune CNS and neuromuscular diseases [1]. The discovery of novel autoantibodies in clinically distinctive diseases has prompted the development of new or revised clinical diagnostic criteria according to the serostatus and new treatment algorithms based on antibody-mediated pathogenesis. Technological developments could increase the detection rate of autoantibodies and help identifying novel pathogenic antibodies in patients who have been regarded as seronegative, which will stratify the diseases in the future. Here, we reviewed the recent discovery of autoantibodies in the CNS and neuromuscular disorders and described their impact on clinical practice.
TYPES OF AUTOANTIBODIES
B cells stimulated by activated T helper cells produce antibodies. Disruption of immune tolerance induces B cells to develop antibodies against self-antigens [2]. These autoantibodies can be classified according to the cellular location of the target antigens. Some of autoimmune diseases have antibodies targeting intracellular proteins, such as nuclear DNA or transcription factors; T-cells could play a central role in the pathogenesis of those diseases. Antibodies targeting extracellular proteins such as ion channels and receptors are usually pathogenic and can serve as diagnostic markers or therapeutic targets [3]. Antibodies binding to neuronal or glial cell surface antigens such as aquaporin-4 (AQP4) or leucine-rich glioma inactivated protein 1 (LGI1) are pathogenic by inducing of inflammation at binding sites or blocking of neural transmission; these neuronal or glial surface antigen-specific autoantibodies play important roles as potential therapeutic targets as well as biomarkers for diagnosis [1,4].
Pathogenic mechanisms of autoantibodies can be affected by the immunoglobulin isotype and subclass of autoantibodies [4]. Autoantibody immunoglobulin G (IgG) subclasses 1 and 3 often cause specific tissue injuries by complement or immune cell activation, but IgG subclass 4 (IgG4) cannot activate the classic complement pathway or activate immune cells [5]. The pathogenic mechanisms of IgG4 autoantibodies are usually different from those of IgG1 or 3 autoantibodies; the pathogenicity of IgG4 is related to the blockade of enzymatic activity or protein-protein interactions of the target antigen [6]. It is known that several antigens targeted by IgG4 autoantibodies are distributed in the CNS or peripheral nervous system [5]; representative IgG4 subclass autoantibodies of neurological disorders include muscle-specific kinase antibody (MuSK-Ab) and neurofascin 155 antibody (NF155-Ab). Table 1 summarizes the various types of autoantibodies and their target antigens in neurological disorders.

Autoantibodies in central nervous system and neuromuscular autoimmune disorders
DETECTION OF AUTOANTIBODIES
To detect autoantibodies, binding of a patient’s antibody to its target antigen is essential; the target antigen can be presented through tissue-based, protein-based, or cell-based assays (Fig. 1) [7]. Enzyme-linked immunoassay (ELISA) using patient sera is a common tool for detecting autoantibodies in routine clinical practice. The size and conformational structure of the protein may limit the accuracy of this method. Immunoassays using radioisotopes have similar limitations. Tissue-based immunohistochemical staining of animal brains is useful for the screening of unknown antibodies. Cell-based immunocytochemistry assay (CBA) and flow immunoprecipitation assay (FIPA) using cells overexpressing specific proteins are powerful tests with high sensitivity and specificity to detect antibodies to specific known antigens. Recently identified autoantibodies of neurological disorders with pathogenic potential usually target cell surface proteins [1]. Therefore, CBA is widely used for the detection of these autoantibodies and can maintain conformational epitopes through transfected cells expressing natively folded proteins [7]. The CBA can be carried out using either indirect immunofluorescence microscopy (IIF) or flow cytometry methods; CBA using IIF is widely used but is semi-quantitative, but CBA by flow cytometry is an automated method with quantification that reduces human bias [8]. And when the CBA is performed in-house, it requires the genetic information of antigenic epitope, transfected cell culture, and skilled analysis of cytochemical staining results, all of which affect the accuracy of the test and limit its application in clinical diagnosis. Commercial kits using prefixed transfected cells can be available in routine clinical practice, but they are usually expensive and require expertise with the interpretation of IIF assays. Multicenter studies that compare the sensitivity, specificity, and reproducibility of antibody assays are ongoing to improve the international standardization of assays [9,10]. These efforts to improve the sensitivity and specificity of assays are important and could facilitate the understanding of the diseases and change clinical practice; a good example of CBA that has reestablished the clinical implications of autoantibodies is the myelin oligodendrocyte glycoprotein antibody (MOG-Ab) [11]. Previous studies of MOG-Ab detected by ELISA and Western blotting showed inconsistent results in patients with multiple sclerosis (MS) and controls. However, CBA with full-length human MOG and IgG1- or IgG Fc-specific secondary antibodies enables the detection of conformation-sensitive MOG-Ab to identify a unique spectrum of diseases; MOG antibody-associated disease (MOGAD) is now considered a CNS inflammatory disease entity distinct from neuromyelitis optica spectrum disorder (NMOSD) or MS [11,12].
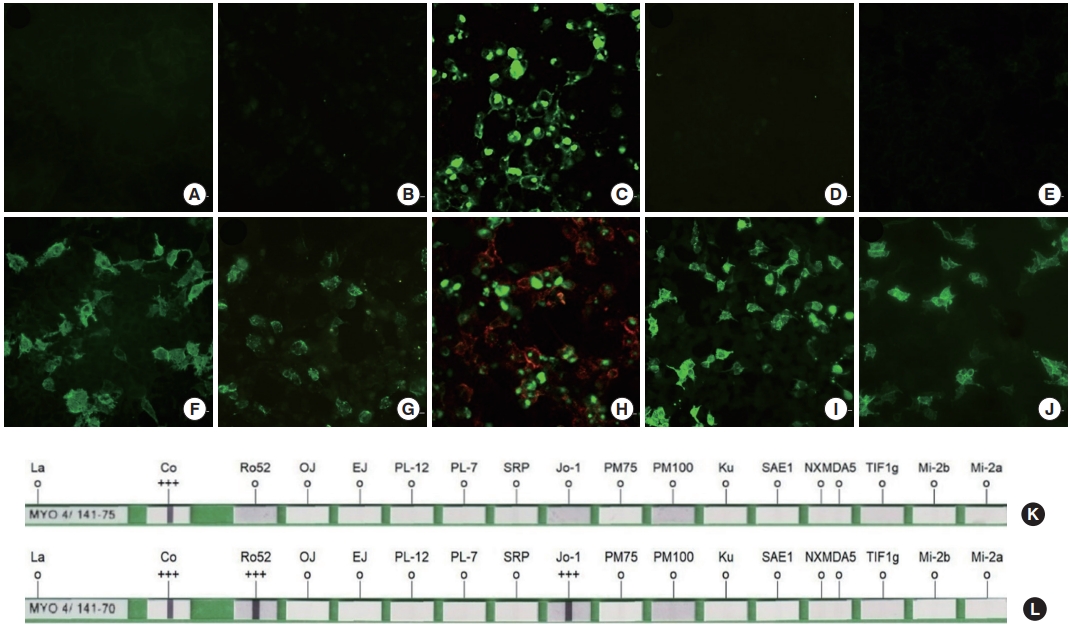
(A-J) Examples of cell-based assays and immunoblots. Representative fluorescence microscopic images of cell-based assays: antiaquaporin-4 antibody (AQP4-Ab), anti-myelin oligodendrocyte glycoprotein antibody (MOG-Ab), and anti-neurofascin 155 antibody (NF155-Ab) in-house cell-based assays and anti-N-methly-d-aspartate receptor antibody (NMDAR-Ab) and anti-leucine-rich glioma inactivated 1 antibody (LGI1-Ab) commercial cell-based assays show negative results in the upper row (A, B, C, D, E) and positive results in the lower row (F, G, H, I, J). Sera with autoantibodies showed the binding of anti-human immunoglobulin G (IgG) antibody (green fluorescence) indicating positive results. In the NF155-Ab in-house assays (C& H), both green and red fluorescence by green fluorescent protein (GFP)-labeled NF155 and Alexa 657 conjugated anti-human IgG antibody were observed in positive sera. (K, L) The commercial immunoblotting for myositis-specific antibodies. The negative sample reacted only in the control region (K), but the positive sample showed binding to specific antigenic sites on the immunoblot strip, followed by the reaction of the enzyme conjugate catalyzing color (L).
AUTOANTIBODIES IN CNS INFLAMMATORY DISORDERS
Multiple sclerosis
MS is a rare disease in Korea and is the most common cause of CNS inflammatory diseases in Western countries [13]. Its pathogenesis is complex, and the mechanism underlying the triggering or progression of the disease is not fully understood. Clinical and experimental evidences indicate the autoimmune pathogenesis of MS; however, an MS-specific autoantibody has not been identified [14]. Pathological studies showed IgG and complement deposition in brain lesions in some cases of MS, suggesting the contribution of humoral immunity and autoantibodies in the pathogenesis [15]; some investigators reported that a subset of patients with MS had autoantibodies inducing demyelinating responses [16,17]. However, screening of autoantigen candidates in MS using advanced tools for peptide and protein analyses has demonstrated no common target antigens, suggesting heterogeneity of the pathomechanisms of MS [17,18]. Although no single autoantibody in MS has been demonstrated, the search for autoantibodies has identified distinct CNS inflammatory disorders characterized by specific autoantibodies such as AQP4 antibody (AQP4-Ab) and MOG-Ab.
T cells have been considered as major players in the pathogenesis of MS; animal models of MS have shown that CD4+ T cells play an important role in experimental autoimmune encephalitis (EAE); for example, interleukin 17 (IL-17)-secreting T helper 17 (Th17) cells dysregulate the blood-brain barrier (BBB) and stimulate the inflammatory process within the CNS [19]. Thus, in terms of treatment, disease-modifying therapies for MS mainly target T cell immunity. Interferon-beta and glatiramer acetate promote anti-inflammatory Th2 cytokines and increase T regulatory cells [20]. Natalizumab, an α4 integrin monoclonal antibody, prevents immune cells in the blood from crossing the BBB [20]. However, with growing evidence from experimental and human pathologies and the success of clinical trials targeting B cells in MS patients, the importance of antibody-dependent or antibody-independent functions of B cells in MS has drawn attention [21]. B cells interact with T cells and promote the secretion of proinflammatory cytokines; conversely, B cells may produce unidentified autoantibodies or inflammatory cytokines. In addition, oligoclonal IgG bands (OCB) in the cerebrospinal fluid (CSF) of MS patients arise from clonally expanded B cells [22]. Antibodies of OCB bind to certain virus/viral proteins or various intracellular proteins that might have been released during tissue injury [23-26], suggesting the pathogenic roles of B cells through humoral immunity in MS [22]. Anti-CD20 monoclonal antibodies, such as rituximab, ocrelizumab, and ofatumumab, have demonstrated excellent efficacy in reducing relapses and progression in relapsing-remitting multiple sclerosis (RRMS). B cells are now considered to play primary roles in the development and progression of the disease. And B cell-depleting therapies are becoming the mainstream treatment in MS [27].
Neuromyelitis optica spectrum disorder
Neuromyelitis optica (NMO) or Devic’s disease was originally described as monophasic simultaneous optic neuritis (ON) and transverse myelitis. Relapsing NMO with brain/brainstem involvement, which is more prevalent in Asian than that in Western countries and formerly known as Asian-type MS, was difficult to differentiate clinically from RRMS. In 2004, NMOspecific IgG (NMO-IgG) from patients with NMO was discovered using tissue-based IIF assays [28]. Subsequent studies have demonstrated that NMO-IgG is pathogenic and directed against a water channel, AQP4, that is abundantly expressed in astrocyte foot processes [29]; this autoantibody was detected in NMO patient sera with high sensitivity and specificity. The differentiation of the disease according to the presence of autoantibodies facilitated the understanding of the disease, and many studies have shown the diversity of clinical and magnetic resonance imaging (MRI) characteristics in patients with NMO [30]. The term NMOSD encompassing these clinical diversities of NMO was introduced in 2007 [31]. Distinctive brain syndromes of NMOSD include the area postrema syndrome, acute brainstem syndrome, acute narcolepsy/diencephalic syndrome, and symptomatic cerebral syndrome. The locations of brain lesions suggest the pathogenetic implications of AQP4-Ab; AQP4-Ab needs to gain access to AQP4 through disrupted/permeable BBB; therefore, preferential locations of brain lesions in NMOSD include circumventricular organs characterized by a lack of BBB, such as the area postrema [32]. In 2015, the international consensus diagnostic criteria for NMOSD were proposed and stratified into AQP4-Ab-seropositive and AQP4-Ab-seronegative NMOSDs, emphasizing the importance of the presence of AQP4-Ab in its diagnosis [33].
The clinical and radiologic features of AQP4-Ab-seronegative NMOSD are mostly similar to those of AQP4-Ab-seropositive NMOSD. But MOG-Ab is detected in some patients fulfilling the criteria of AQP4-Ab-seronegative NMOSD, so this disease category appears to be heterogeneous in nature [34]. AQP4s in astrocytes are expressed by alternatively spliced transcripts encoding two major isoforms, M1 and M23 [35]. M23-AQP4 assembled in membranes display a regular square distribution called orthogonal arrays of particles (OAPs), which have a higher affinity for AQP4-Ab than that of M1- AQP4, which does not form OAP. In fact, the AQP4-Ab binding assay with M23-AQP4 improved the detection sensitivity from 70% to 97% for NMO-IgG derived from human serum samples of NMOSD [36]. These M23-AQP4-binding NMO-IgGs are effectively detected by several experimental methods, such as ELISA and CBA, which are more sensitive than animal brain tissue-based IIF. Moreover, based on a systematic review by Waters et al. [37], the live cell-based assay (76.5%) was more sensitive for AQP4-Ab detection than that in ELISA (61.8%), IIF assays (61.2%), radioimmunoprecipitation assay (55.2%), and FIPA (48.5%). The specificity of CBA (99.8%) was also higher than that of other detection methods, such as ELISA (96.6%) or IIF assays (97.4%). Cell lines like human embryonic kidney cells (HEK293) transiently transfected with human AQP4 express conformationally folded AQP4 on the cell membrane, which enables CBA to be superior to ELISA using linear peptides [10,37]. In pursuit of identifying antibodies from seronegative NMOSD, MOG-Ab particularly attracted attention; up to 40% of patients with AQP4-Ab-seronegative NMOSD may show MOG-Ab positivity [38]. Currently, MOGAD is considered a disease different from NMOSD. We have gained a deeper understanding of NMOSD with the discovery and improvement of autoantibody testing, and recently new treatment options for NMOSD have received U.S. Food and Drug Administration approval. Eculizumab (anti-C5), inebilizumab (anti-CD19), and satralizumab (anti-IL-6 receptor) showed high efficacy in preventing relapses in AQP4-Ab-NMOSD. Thus, a new treatment era for NMOSD is emerging, although therapy of AQP4-Ab-seronegative NMOSD remains a challenge.
Myelin oligodendrocyte glycoprotein antibody-associated disease
MOGAD has recently been recognized as a distinct clinical entity with the detection of MOG-Ab, which encompasses variable clinical phenotypes, including ON, acute disseminated encephalomyelitis (ADEM), AQP4-Ab-negative NMOSD, encephalitis, myelitis, and brainstem encephalitis [39]. MOG itself has long been considered a target antigen in CNS demyelinating diseases including MS and ADEM because MOG is localized on the surface of the myelin sheath, and EAE animal model for MS can be induced by MOG peptides and MOG-reactive T cells. However, the clinical implications of MOG-Ab in MS were unclear when MOG-Ab was assayed through ELISA and Western blotting. Since Mader et al. [40] and O’Connor et al. [41] found conformation-sensitive MOG-Ab in patients with certain clinical syndromes of CNS inflammatory disorders, research is ongoing to understand the pathogenic roles of MOG-Ab and develop more accurate detection methods. A multicenter comparison study showed that MOG-Ab CBA was an effective method [42]. The CBA using live cells transfected with full-length human MOG and anti-IgG1 as secondary antibody showed high specificity for non-MS demyelinating diseases [43]. A meta-analysis revealed that MOGAD represented 9.3% of NMOSD and 32.5% of AQP4-Ab-seronegative NMOSD [44]. The clinical and imaging characteristics of MOGAD overlap partly with those of MS and AQP4-Ab-seropositive NMOSD, whereas features distinguishing MOGAD from MS or NMOSD can also be determined [45]. A recent pathological study showed that perivenous demyelination with MOG-dominant myelin loss was a distinctive finding of MOGAD, which is apparently different from that of MS and NMOSD [46]. For example, chronic recurrent isolated optic neuritis (CRION) with perineural gadolinium enhancement on orbital MRI, conus medullaris involvement in myelitis, and ADEM in children are common features of MOGAD. Natural courses and treatment algorithms have not yet been established for MOGAD. The response to high-dose corticosteroid treatment at the time of attack is relatively good, but the efficacy of immunosuppressive agents to prevent relapses remains to be determined [47]. Several questions remain to be answered in MOGAD, but MOGAD is now considered an independent autoimmune CNS demyelinating disease, and testing for MOG-Ab in patients suspected of having CNS demyelinating diseases is essential [48].
N-methyl-D-aspartic acid receptor antibody and leucine-rich glioma-inactivated 1 antibody in autoimmune encephalitis
With the discovery of autoantibodies, new forms of autoimmune encephalitis have been identified; these encephalitides manifest some clinical characteristics similar to those in infectious or paraneoplastic limbic encephalitis, but also have distinctive features associated with specific autoantibodies against neuronal cell surface or synaptic proteins [49]. N-methyl-d-aspartate receptor (NMDAR) encephalitis is a common type of autoimmune encephalitis. Psychosis, memory deficits, and seizures occur initially, and then the disease progresses to a later stage of unresponsiveness with catatonia and autonomic instability [50]. Most patients with NMDAR encephalitis are women. Neoplasms, especially ovarian teratoma, are known to be associated with the disease [51]. Pathological studies of the brain and coexisting tumors in cases of NMDAR encephalitis revealed that all these tumors had NMDAR-expressing neuronal components, suggesting that the tumor could trigger the generation of NMDAR antibody [52]. Although IgG subclasses of NMDAR antibodies are IgG1 and IgG3, complement deposition was not detected in the brains of patients with NMDAR encephalitis [52,53]. This suggests that this autoantibody does not induce complement-mediated cytotoxicity, but a decrease in synaptic NMDAR expression by crosslinking and internalization could be a pathogenic mechanism related to the autoantibody [51-54].
LGI1 encephalitis is another clinically recognizable autoimmune encephalitis; faciobrachial dystonic seizures precede the development of memory disturbance or confusion that characterizes limbic encephalitis [55]. LGI1 is a cell surface protein complexed with voltage-gated potassium channels (VGKCs). Other VGKC-complex antibodies, except the LGI1 and contactin-associated protein-like 2 (CASPR2) antibodies, bind intracellular components, lack pathogenic potentials, and are not related to specific syndromes [56,57].
With these pathogenic autoantibodies related to specific types of autoimmune encephalitis, the treatment strategy of early active immunotherapy in autoimmune encephalitis is further specified and now recommended [58]. Autoantibody tests are commercially available, and using both CSF and serum for the tests is recommended due to the lower background activity and higher specificity with CSF, but the LGI1 antibody can be preferentially detected in serum [57].
AUTOANTIBODIES IN NEUROMUSCULAR DISORDERS
Neurofascin 155, contactin-1, and contactin-associated protein 1 antibodies in chronic inflammatory demyelinating polyradiculoneuropathy
Chronic inflammatory demyelinating polyradiculoneuropathy (CIDP) is a chronic immune-mediated peripheral nerve disorder and is the most common acquired inflammatory neuropathy; however, CIDP is clinically heterogeneous. Typical CIDP presents with symmetrical sensory and motor dysfunction, a recurrent course, and response to steroid or immunoglobulin treatment; there are also atypical forms of CIDP (CIDP variants), including distal dominant demyelination, focal or multifocal, and pure motor or sensory types [59]. Chronic demyelinating neuropathy is often accompanied by dysproteinemia, such as monoclonal gammopathy of unknown significance (MGUS), or hematologic malignancies such as multiple myeloma or polyneuropathy, organomegaly, endocrinopathy, M-protein, and skin changes (POEMS) syndrome. Because of heterogeneous clinical features and treatment response, CIDP is frequently misdiagnosed [60]. Classification based on pathomechanisms, biomarkers for early diagnosis, and personalized strategies of therapeutic algorithms are still unmet needs. Earlier efforts to identify disease-specific autoantibodies in CIDP failed, but autoantibodies have been found in other chronic immune-mediated neuropathies; anti-ganglioside GM1 IgM antibody is found in approximately half of patients with multifocal motor neuropathy (MMN), and anti-myelin-associated glycoprotein (MAG) IgM antibodies are frequently detected in distal dominant acquired demyelinating symmetric neuropathy accompanying IgM-MGUS [61]. Autoantibodies could play an important role in the pathogenesis of CIDP, at least in some patients [62].
Recent studies have identified autoantibodies against nodal/paranodal proteins in a subset of patients fulfilling the 2010 diagnostic criteria for CIDP. Screening of target antigens of IgG through proteomic analysis and mass spectroscopy, and detection of autoantibodies from the sera of patients through ELISA and CBA identified autoantibodies against neurofascin splice variants 155 (NF155) and 186 (NF186), followed by contactin‑1 (CNTN1) and Caspr1 [63,64]. NF155, CNTN1, and Caspr1 are cell adhesion molecules in the paranodal junction of the Ranvier node; NF155 on the myelin side of the paranodal junction interacts with CNTN1, which forms a complex with CASPR1 on the axonal side [65,66]. This nodal/paranodal complex in the paranodal junction is important for saltatory conduction by blocking nodal currents into the internode [66]. A meta-analysis found a high variability in the frequency of the autoantibody detection with a very low diagnostic sensitivity (9.0% in anti-NF155 antibodies) and a high specificity [67]. A highly specific autoantibody detection test is important for diagnostic utility, and CBA and ELISA are commonly used, but ELISA may be associated with more false-positive and false-negative results [61].
Autoantibodies against NF155, CNTN1, and Caspr1 are predominantly IgG4 isotypes. CIDP with autoantibodies directed against paranodal junctions differs pathogenetically from typical CIDP; the traditional concept of CIDP pathogenesis represented that T-cells, macrophages and complements work together to induce the disease [68,69]. However, CIDP with autoantibodies shows IgG4 deposition without macrophage infiltration, and axonal damage is probably due to sodium channel dysfunction and disturbance of glial support to axons. One study using electron microscopy revealed that detachment of terminal myelin loops from the axolemma at the paranode was frequently found in CIDP with NF155 and CNTN1 antibodies [70]. Besides the difference in pathogenesis, CIDP with autoantibodies differs clinically from typical seronegative CIDP. Subacute onset and younger age at onset with tremor or ataxia are more frequently observed in NF155-Ab-positive CIDP [65,71]. CNTN1 and Caspr1 antibodies are detected in a small fraction of CIDP cases with severe motor involvement or pain; most of these CIDPs with autoantibodies show poor response to intravenous immunoglobulin (IVIg) therapy. Rituximab, an alternative to IVIg therapy, shows promising treatment results in a portion of refractory CIDP with autoantibodies [72].
Distinct clinical features and IgG4-related pathogenesis make CIDP with autoantibodies a distinct group of diseases, and recent diagnostic and treatment guidelines of CIDP proposed to call these disorders “autoimmune nodopathies” rather than CIDP variants [59]. Currently, it is evident that seropositive CIDP should be differentiated from typical CIDP, and serologic tests to detect paranodal junction autoantibodies are necessary in clinical practice despite the low frequency of seropositivity in CIDP.
Muscle-specific kinase antibody and low-density lipoprotein receptor-related protein 4 antibody in myasthenia gravis
Myasthenia gravis (MG) is an autoimmune neuromuscular junction disorder. With the pathogenic autoantibody against the acetylcholine receptor (AChR), MG is a prototype of antibody-mediated autoimmune disease [73]. AChR antibodies (AChR-Ab) are found in most patients with generalized MG, but approximately 15% of patients are seronegative [74]. Further efforts to find other autoantibodies in seronegative MG focused on the other components of the muscle endplate at the neuromuscular junction, including the MuSK, agrin, and low-density lipoprotein receptor-related protein 4 (LRP4), which can affect the function of neuromuscular junction transmission [75]. Antibodies against MuSK are found in 30% to 50% of AChR-Ab-negative MG patients [76], and MG with MuSK-Ab is clinically different from AChR-Ab-positive MG; they show more severe bulbar symptoms, frequent muscle atrophy, and less prominent fluctuation of weakness and are not associated with thymic disorders [77]. MuSK-Ab is predominantly IgG4 [78]; thus, the main effector mechanism of MuSK-Ab is thought to be related to the inhibition of protein function or interaction rather than complement activation; MuSK and LRP4 interaction is needed to induce dense clustering of AChR by agrin secreted from the axon terminal, which could be interfered by MuSK-Ab [79,80]. LRP4 antibodies are mostly IgG1 and detected in a small percentage of double-seronegative patients for AChR-Ab and MuSK-Ab [81]. The pathogenic roles of the LRP4 antibody have been reported in animal models immunized with LRP4, or the passive transfer of the LRP4 antibody can cause MG-like symptoms [82], but some controversies remain; the LRP4 antibody could be positive in approximately 20% of patients with amyotrophic lateral sclerosis [83].
Radioimmunoassay is traditionally the gold standard in detecting AChR antibodies based on the 125I-α-BuTx-labelled AChR from TE671 cells. The specificity of the test is very high, with a sensitivity of 80% to 85% in generalized MG, and changes in the titer can be monitored quantitatively [84,85]. ELISA, FIPA, and luciferase assays have the benefits of avoiding the hazards of radioactivity, but these methods are not widely used due to lower accuracy or difficulty in routine use [86,87]. CBA using AChR-expressing HEK293 cells showed high sensitivity and specificity; rapsyn need to be co-transfected to promote receptor clustering [74]. Recently, clinical trials using monoclonal antibodies directed against specific targets, including complement, transforming growth factor, or neonatal Fc receptors have been actively conducted. To apply a specific immunotherapy, it has become more important to determine the patient’s serostatus.
Myositis-specific antibodies
Autoantibodies of myositis can now be detected in clinical settings where antibodies were not expected to be present previously. Some of the new autoantibodies are anti-transcriptional intermediary factor 1γ (TIF1γ) antibody, anti-nuclear matrix protein 2 (NXP2) antibody in dermatomyositis, anti-melanoma differentiation-associated gene 5 (MDA5) antibody in myositis with overlapping features, and anti-signal recognition particle (SRP) and anti-3-hydroxy-3-methylglutaryl CoA reductase (HMGCR) antibodies in immune-mediated necrotizing myositis. Unique clinical features associated with these autoantibodies are observed, which have resulted in a major change in the classification of myositis [88,89]. These autoantibodies were usually detected by immunoprecipitation, which could be considered as the gold standard test; however, there is a limitation on the use of this in clinical practice [90]. These limitations can be overcome by commercial multiplex dot or line immunoblots. However, recent studies using these methods showed variable specificity but low sensitivity with large differences in diagnostic performance according to clinical settings [90-92]. Recent classification criteria for idiopathic inflammatory myopathies include autoantibodies for classification; the role of autoantibodies in inflammatory myopathies is growing [93]. However, their use for the diagnosis of inflammatory myositis is still a challenge, and further validation of these assays is needed [94].
CONCLUSION
The clinical approach to autoimmune diseases has significantly changed in the past decades with a better understanding of the pathogenesis for which the discovery of autoantibodies plays a crucial role. Newly discovered autoantibodies have provided a better insight into the pathological mechanisms of these diseases and contribute to the development of specific therapies. Autoantibody testing is currently mandatory for clinical practice because clinical features and treatment strategies for seropositive diseases are different from those of seronegative diseases. However, limitations in the availability of new antibody testing methods for physicians still exist. CBA is a useful method to identify autoantibodies with high sensitivity and specificity, but it is difficult to establish and maintain in-house cells for CBA in terms of technical and cost aspects. Commercial CBA kits using fixed cells are widely used in hospital laboratories or outsourced, but their use is limited due to high test costs, and caution is required in the interpretation of results. Continuing refinement of autoantibody testing through research is required to discover new target antigens or to be applied in clinical use.
Since most CNS and neuromuscular autoimmune diseases are rare diseases, multicenter cooperative research is imperative for the standardization of test methods and accumulation of clinical data. In response to this need, a Korean research network for Risk factor analysis of Autoimmune diseases in the Central nervous system (KoRAC) was established to support antibody testing and collect prospective clinical data and biological samples. It is expected that such a multicenter study will take a step closer to the development of more efficient diagnostic methods and treatments.
Notes
Mi Yong Jeon and Jin Myung Seok have no conflicts of interest to declare. Byoung Joon Kim has received honoraria and/or consulting fees from Bayer, Genzyme, Merck, Celltrion, Astellas, Genuv, and Corestem. Kazuo Fujihara serves as an advisor or on scientific advisory boards for Biogen, Mitsubishi Tanabe, Novartis, Chugai/Roche, Alexion, Viela Bio/Horizon Therapeutics, UCB, Merck Biopharma, Japan Tobacco, and AbbVie; has received funding for travel and speaker honoraria from Biogen, Eisai, Mitsubishi Tanabe, Novartis, Chugai, Roche, Alexion, Viela Bio, Teijin, Asahi Kasei Medical, Merck, and Takeda; and has received the Grants-in-Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science and Technology of Japan and the Grants-in-Aid for Scientific Research from the Ministry of Health, Welfare and Labor of Japan.
AUTHOR CONTRIBUTIONS
Conception or design: JMS, BJK.
Acquisition, analysis, or interpretation of data: MYJ, JMS.
Drafting the work or revising: MYJ, JMS, KF, BJK.
Final approval of the manuscript: MYJ, JMS, BJK.
Acknowledgements
This study was supported by the Korean Centers for Disease Control and Prevention (no. 2017E6300202).