Bioinformatics challenges in molecular epidemiology of cancers
Article information
Abstract
Molecular epidemiology is the integration of molecular biologic techniques into epidemiologic study. With the advances in understanding of carcinogenesis and the human genome, there has been an evolution in the field of cancer epidemiology. However, traditional analyses of single genetic variants often fail to identify susceptibility genes for cancer risk. In particular, recent technological evolution has enabled high-throughput analyses for a number of genetic variants and driven accumulation of unprecedentedly large genome data, imposing bioinformatics challenges. These studies aim to integrate the genetic basis of complex diseases including cancers in which the interplay of multiple genetic and environmental risk factors may play an important role. Here we outline currently available approaches for detecting variants of cancer risk. We also review upcoming bioinformatics challenges and technical aspects in the field of molecular epidemiology, and discuss their future impact on the understanding of carcinogenesis and personalized strategies for cancer prevention and therapy.
INTRODUCTION
Epidemiology is the study of sick and well people to determine the crucial difference between those who get disease and those who are spared, and has been very successful in identifying environmental factors that modify the risk of cancers, leading to cancer prevention strategies. Traditional epidemiology is concerned with correlating exposures with cancer incidence, using the Bradford-Hill criteria for assigning causality to an association in which a specific exposure might cause a specific cancer type [1]. However, the etiology of many types of cancers is still poorly understood, despite extensive use of questionnaires and interview-based approaches in conventional epidemiologic studies. For example, in certain areas of the world where Helicobacter pylori infection is prevalent, only a small fraction of those infected develop gastric cancer. Therefore, it has been widely recognized that not all individuals respond in the same manner to similar pharmaceutical or environmental exposures. Limited knowledge of interindividual variability in response to exposures based on inherited variants results in the need for rational integration of molecular analysis into cancer epidemiologic studies.
Susceptibility to environmental exposures
During the past decade, epidemiologic research has undergone major technological and methodological development, with incorporation of molecular, cellular, and other biologic measurements into traditional approach [2]. The etiology of cancers involves strong environmental and genetic components, in which the global distribution is characterized by a wide geographic variation in incidence. In molecular epidemiology, the epidemiologist is much more of a participant in the assessment of the biologic basis for an association, by using biologic measurements to assess exposure, internal dose of carcinogen, early biologic effect resulting in altered structure and function, and outcome. Furthermore, interindividual differences in the absorption, activation, and detoxification of carcinogens, or the response to DNA damage caused by carcinogens, may mediate the relation between exposure and outcome [3]. The most common markers of the susceptibility are mutations in specific genes that confer increased or decreased risk. Familial clustering of specific cancer sites has long been recognized, and family history of a specific cancer is associated with increased risk. For example, hereditary nonpolyposis colorectal cancer (HNPCC) syndrome is associated with mutations in the mismatch repair (MMR) genes [4]. Subsequent analyses of a larger set of families showed that germline mutations in the MMR genes are responsible for 70% to 90% of all HNPCC cases [5].
Almost all carcinogens require activation induced by metabolic enzymes, and detoxification enzymes frequently exist to deactivate carcinogens or their intermediate metabolites. Inherited polymorphisms in these enzymes may alter their rate of activation or detoxification; thus, increasing or decreasing the carcinogenic potential of the environmental exposures they act on. Furthermore, as our knowledge of the molecular mechanisms of carcinogenesis has expanded rapidly, gene pathways have become new focus of molecular epidemiology research. These include pathways involving DNA repair, cell cycle control, immune response, and the inflammatory response. Genes in these pathways are candidates in the search for interindividual genetic variants that may modify cancer risk. Of note, the completion of the Human Genome Project naturally led us to examine the genetic variations that presumably underlie the fact that a family history of cancer is a major risk factor for most cancer types [6,7]. These genes may also interact with environmental factors such that cancer risk is not equally elevated in all persons exposed to a carcinogen or all gene carriers.
Determination of treatment outcome
One of the limitations in cancer treatment is the recognition that some patients suffer toxicity from chemotherapy that may be fatal, compromise quality of life, or limit the dose of drugs able to given. The variation in drug tolerance is thought to be results from inherited differences in drug metabolism. Likewise, some patients respond to certain chemotherapeutic agents, while others experience disease progression or toxic reactions. In fact, the application of pharmacogenetic/genomics to cancer treatment outcomes is the area that holds exceptional promise. In studies of cancer etiology or susceptibility, most of the variants identified for cancer risk infer risks that are slight, and it is likely that many frequent (>1%) single nucleotide polymorphisms (SNPs) will not increase risk of cancer but will only become penetrant in the presence of exposures that are relevant for disease etiology. However, for studies of treatment outcomes, the exposure is known and common to all individuals receiving treatment, yet not all experience the same toxicities. One of the best examples of inherited differences in chemotherapy outcome is the pharmacogenomic study of oxaliplatin-induced severe neuropathy [8]. About one-third of patients treated with oxaliplatin experienced severe neuropathy [9]. Genome-wide association (GWA) analyses found five polymorphisms (rs10486003, rs2338, rs830884, rs843748, and rs797519) that could be associated with the toxicity. Theoretically, screening for these SNPs may not only avoid the risk of severe toxicity, but may also help optimize chemotherapy regimen and dosage.
DETECTION OF GENETIC VARIANTS
Candidate gene studies have provided valuable data in the areas of pharmacogenetics and pharmacogenomics. This is especially the case for adverse drug reactions attributable to alleles of a single gene, which often encodes an enzyme contributing to metabolism of the drug. However, the availability of GWA approaches enabled contributions from novel and less obvious genes to be detected, especially in the area of susceptibility studies, which is more complex and less well understood than the pharmacogenetics of drug metabolism. Currently, there is considerable interest in applying whole-genome sequencing to molecular epidemiology with a view to identifying rare genetic variants. This may involve sequencing of all coding regions (exome sequencing) or entire genomes using the new technologies that are rapidly developing [10]. Owing to highly coordinated efforts including the HapMap Project and the 1000 Genomes Project [7,11,12], discovery of genetic variants has become one of the most interesting areas in which many procedures require complex and highly demanding computation [13].
Gene-gene or gene-environmental interactions
The current strategy for revealing the genetic basis of disease susceptibility is based on the common disease common variant hypothesis [14], supporting that genetic variations with alleles that are common in the population will explain the genetic basis of common disease. That is, if a variant is distributed non-randomly with a disease, it could be linked to a susceptibility gene. Our goal in molecular epidemiology is to understand the relationship between individual variation in DNA sequences, in environmental exposure and in disease susceptibility, resulting in the improvement of diagnosis, prevention, and diagnosis. Unfortunately, despite GWA studies have identified a number of common SNPs, the identified variants explain only a small proportion of the heritability of complex diseases [15], which implies that multiple SNPs or environmental risk factors may contribute to disease susceptibility but individual factors confer only modest contributions to disease risk. Given the failure to identify novel susceptibility genes using GWA studies indicates limitations of this approach, and the major technological advances enabling high-throughput genotyping or sequencing, there has been the emerging need to shift from the single SNP approach towards a more holistic one in order to recognize the complexity of the gene-gene and gene-environmental interactions. For example, we all know that only a few individuals infected by H. pylori develop gastric cancer [16], even in areas with a high incidence of H. pylori infection.
Considering the non-linearity between the genotype and phenotype, it can arise from phenomena such as genetic heterogeneity (i.e., different DNA sequences leading to the same phenotype), phenocopy (i.e., environmentally determined phenotype without a genetic basis) and the gene-gene or gene-environmental interactions [17]. The value of studying gene-environmental interactions may be found in that it is allowed to refine risk estimates associated with specific exposures, by focusing primarily on those who are most susceptible and at risk. While association between cancer risk and specific exposure may be weak or absent in a heterogeneous population, association may be found in the examination of only those who are most susceptible.
“Big data” problem
With the tremendous progress in the technology in genomics, there has been an exponential growth in the amount of data, which has been widely recognized to be a critical barrier in analyses and interpretation. For instance, the amount of data that have been archived in the NCBI database for the past two years already exceeded the total amount that had been archived before that. Even before analysis step, there happen immediate challenges regarding data storage and exchange among data generating groups and analyzing groups. Such a huge scale of data is far beyond computational capacity in most biomedical research labs. To overcome this practical difficulty for building a server for data storage and extensive computation in individual labs, many companies such as Amazon have developed cloud computing platform and provided service to allow researchers use their servers as needed or many research institutes have built clustering computing platform for their affiliated labs.
On the other hand, the majority of biologic or clinical researchers are still unfamiliar with computational approaches to deal with those data, and bioinformatics is a foreign territory. Recent microarray or sequencing-based experiments often generate substantially bigger data and are more broadly applicable than before [18], and the problem may occur when most biomedical researchers have very limited capacity to carry out analyses of such big data using appropriate tools that can be fully understood by others. To study the effects of genetic variants on cancer risk, therefore, involvement of computational biologists and bioinformaticians with expertise and knowledge in genomics and computational domains becomes more unavoidable and impending. In addition, considering gene-gene or gene-environmental interactions as important factors for cancer susceptibility, as well as markers other than genetic polymorphisms including copy number variation, mitochondrial DNA, variations in microRNAs and other factors that regulate gene expression, there is no currently available ideal method to deal with such diverse large datasets.
STUDY DESIGNS FOR MOLECULAR EPIDEMIOLOGY
Current focus is on integrating findings from the large sets of data that have been generated through large consortia. Studies of gene-environmental interactions should require careful consideration of epidemiologic study design, exposure assessment and methods of analysis, with particular attention to data quality [19]. Integration of GWA data with expert biological knowledge would be equally important.
Epidemiologic study designs include observational (e.g., cohort, case-control, or case-only) studies that do not involve any intervention or experiment, and experimental studies that entail manipulation of the exposure and randomization of subjects to exposure groups (Table 1). The issues for choosing appropriate one among different designs include the control of confounding factors and other sources of bias, the temporal relationship between exposure and disease, data quality, the ability to test multiple endpoints, and the efficiency of detecting rare diseases or rare risk factors. One of the major challenges to the success of epidemiologic studies is that the uncertainties in exposure assessment can lead to unpredictable biases, especially if they differ with respect to disease, as well as induce spurious interactions. Moreover, enough sample size required for most epidemiologic studies can be enormous. Thousands of cases are typically required for case-control studies, and even tens of thousands are needed in GWA studies because a more stringent significance level is required at the genome scale [20].
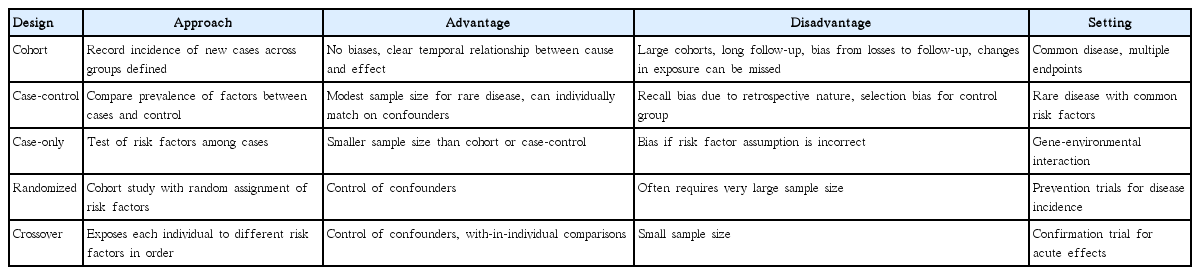
Designs for epidemiologic studies
Although any of the standard epidemiologic designs for studying effects of genes or environmental factors can be applied to the study of gene-environmental interactions (Table 2), the computational burden and general absence of prior knowledge about most SNPs result in additional problems. Conventional analyses of GWA data often involve exhaustive scan for all possible pair-wise interactions but can still miss those with weak marginal effects [21]. In addition, scanning for higher-order (i.e., gene-gene-gene or gene-gene-environmental) interactions is computationally less feasible without filtering based on lower-order interactions. Another challenge that deserves to be mentioned still remains in the biological interpretation of non-linear genetic models. While a computational model can be made to identify SNPs that increase susceptibility to disease, the specific mathematical relationships cannot be translated into diagnosis or treatment strategies without interpreting the results in the context of human biology.
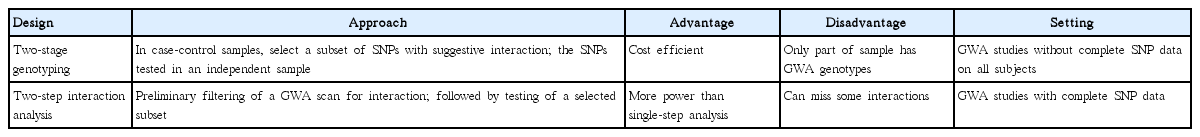
Designs for gene-environmental interactions
BIOINFORMATICS IN MOLECULAR EPIDEMIOLOGY
Previous association studies mainly focused on genetic factors at a time only taking a limited number of demographic variables such as age, gender, ethnicity etc. as covariates without fully considering the environmental complexity of disease. In addition to the failure to identify relevant susceptibility genes using high-throughput genotyping or GWA studies [15], as the technology continues to change rapidly, there is a challenge arising due to not only too little knowledge but also too much information. In mapping relationship between interindividual variations in DNA sequences, environmental exposures and disease susceptibility, one should consider the amount of non-linearity in effects of a genotype or phenotype [17]. In addition to large genomics data, various omics data including transcriptome, proteome, metabolome etc. will not only provide valuable opportunities to understand human cancers systematically but also raise more stubborn challenges. The Cancer Genome Atlas Project has shown great success of integrative approaches in dealing with such opportunities and challenges [22]. To address the complexity of the underlying genetic basis of disease, and to deal with unbelievably large amount and diversity of data including gene-gene or gene-environmental interactions, bioinformatics can play an important role.
Data mining
Considering the complex biologic phenomena such as gene-gene or gene-environmental interactions will make up much of the genetic basis of disease, computational modeling can be very challenging because the combinatorial number of genotypes and gene-environmental combinations goes up exponentially as more variable is added to the model. Many exploratory methods have been developed for multivariate analysis of high-dimensional data, ranging from standard multiple regression methods to a number of data mining or machine learning techniques [21], but traditional linear, parametric statistical approaches have limited power for modeling high-order, non-linear interactions that are likely to be important in the etiology of complex diseases. The advantage of data mining methods is that they are based on fewer assumptions and thus less biased about the functional form of the model and the effects being modeled [23].
One of the most popular machine learning algorithms for studying interactions is random forest (RF). In RF, variables such as SNPs, environmental and/or demographic factors are designated as attributes. RF is often used for selecting the subset of attributes that can be modeled using decision trees. Decision trees are used for modeling the relationship between one or more attributes and an endpoint, leading to classification of subjects as case or control by sorting them through a tree from node to node where each node is an attribute with a decision rule. A RF is a collection of individual decision trees, where each tree in the forest has been trained using a bootstrap sample of subjects from the data, and each attribute in the tree is chosen from a random subset of attributes [24]. The primary advantage of RF is that it is simple and the resulting tree can be interpreted as a series of if-then-else rules that are easy to understand. Furthermore, RF is a useful approach for studying gene-gene or gene-environmental interactions because importance scores for particular attributes take interactions into account without demanding a predefined model.
Selection or filtering SNPs for combinatorial analysis
A second challenge in human genetics and molecular epidemiology is the selection of SNPs that should be included in the analysis. If non-linear interactions between genes explain a significant proportion of the heritability of cancers, then a number of combinations of SNPs will need to be evaluated from a thousand-to-millions of candidates. It is now commonly understood that at least millions of carefully selected SNPs are necessary to capture the relevant variation across the human genome. With these many attributes the number of higher order combinations is astronomical. Therefore, filtering or selection procedures will play an important role in GWA studies because there are more possible combinations of SNPs to test than can be exhaustively evaluated using modern computational horsepower [25].
To find the optimal number of attributes and to detect unknown important interactions between attributes, there are two approaches to selecting attributes for predictive models. The filter algorithm can process the data by assessing the quality or relevance of each variable and then using the information to select a subset for analysis. A standard statistical strategy in human genetics and molecular epidemiology is to assess the association of each SNP with a disease using a chi-square test of independence followed by a correction of the significance level that takes into account an increased false positive rate due to multiple tests. This is very efficient in assessing the independent effects of SNPs on disease susceptibility but it ignores the interactions between attributes. Algorithms such as Relief, ReliefF, or SURF (Spatially Uniform ReliefF) show promise for filtering interacting SNPs or attributes [23], but still have a limitation that infrequent, but actually important, attributes might be discarded prior to analysis.
On the other hand, the wrapper method iteratively selects subsets of attributes for classification using either a deterministic or stochastic algorithm; thus, may be more powerful than filter method because no attributes are discarded in the process. One of the most advanced methods involving wrapper algorithm is genetic algorithm (GA), a machine learning method to search for optimal rules or combinations that satisfy a predefined condition [26]. In the wrapper approach, the goal of GA is to evolve diverse combinatorial sets of attributes to find optimal interactions in a large number of possible combinations, which can be accomplished by repeating the following steps: first generating or initializing a population of random combinations that are composed of the basic units (SNPs or attributes), evaluating the combinations, and then delivering combinations with high evaluation to next generation. For genetic studies the basic units might be a list of SNPs or environmental factors along with a list of mathematical functions. Each randomly generated combination is evaluated and the good combinations are selected based on fitness. This process of selection and recombination or mutation to generate variability is repeated until best combinations are identified. One caveat is that the best combinations or SNP sets detected in one data should be validated in independent datasets to avoid the possibility of over-fitting of model to the discovery data.
BIOMEDICAL KNOWLEDGE FOR ANALYSIS AND INTERPRETATION
Even when a computational model can be made to identify SNPs or interactions that increase the disease risk, the biological interpretation of the results may be the most important and difficult challenge of all. It will be more apparent when statistically significant SNPs are found in gene desert regions [16,27]. As one of these efforts, the ENCODE (Encyclopedia of DNA elements) project have generated an amount of functional elements in the human genome [28]. A previous study showed that a high portion of disease-associated variants are enriched in functional elements [29], which suggests that availability functional data enhance our ability to interpret statistical findings yet-to-be meaningful. Besides biologic data are often difficult to obtain, there is growing recognition that biomedical knowledge can guide genetic association studies to more meaningful results. For example, for any given disease there are often multiple biochemical pathways that play an important role in disease development. Pathway-based analyses of genetic association studies are more likely to replicate than individual SNPs [30], and the use of prior knowledge about pathways can facilitate the analyses [31]. Web-based tools such as the gene set enrichment analysis and the DAVID (database for annotation, visualization and integrated discovery) have been widely used for pathway enrichment analysis [32,33].
Despite of availability of several well-designed web-based tools, a general drawback of current computational techniques, for biomedical researchers in particular, is the lack of simplicity. There have already been a number of leading-edge analysis tools often distributed through SourceForge, Google Code, and others, including direct downloads from developers’ websites. However, most of these tools are of little use to biomedical researchers because specific skills are often required to compile, install, and use them. The key to successful GWA study is the close collaboration between biomedical researchers, biostaticians, and bioinformaticians.
Gene and pathway-based analysis of GWA data
The common approach to GWA studies is to select tens to hundreds of the most significant SNPs for further investigation. However, as described above, common diseases often arise from the joint action of multiple loci or multiple genes within a pathway. A gene or a pathway consists of a group of interacting components that act in concert to perform specific biologic tasks. Furthermore, the function of many SNPs may not be well characterized yet, but the function of genes and particular pathways have been much better investigated. The gene and pathway-based analysis considers a gene or a pathway as the basic unit of analysis [34,35], with the aim of identifying simultaneous associations of a group of genetic variants in the same gene or biochemical pathway. On the other hand, expression quantitative trait loci analysis identifies statistically significant correlation between each SNPs and gene expression level of particular genes in cis- (locally) or trans- (at a distance) effects [36]. One of the major advantages of studies at the gene or pathway level is that pathway-based analysis can add structure to genomic data and allows us to gain insight into a deeper understanding of the biologic consequences at cellular level as networks of functionally related genes. Integration of gene expression data and DNA variants increases computations exponentially and necessarily induces false positive findings. Another challenge facing us is the fact that the current understanding of human gene function is not complete, and a large number of genes or pathways are still uncharacterized or poorly characterized.
CONCLUSION
Molecular epidemiology has been focusing on dissecting the heterogeneity of susceptibility leading to a better understanding of causes of disease. High-throughput genotyping and GWA studies have generated a number of important bioinformatics challenges including modeling of complex genotype-phenotype relationships using data mining and developing new machine learning methods. As we encounter more and more data and discover new complexities in the human genome, although there has not been a widely accepted theory for how to analyze genomics and other omics data and interpret those results, powerful bioinformatics research strategies would be even more critical for identifying genetic risk factors for human cancers. Successful genetic and molecular epidemiologic study is not only dependent on the quality of big data or expert biologic knowledge, but also on data mining requiring high computational efforts. Given these considerations, we indeed need to interact and collaborate among biologists, translational researchers, and bioinformaticians to tackle emerging important bioinformatics challenges in genetic studies.
Notes
No potential conflict of interest relevant to this article was reported.